RNAflow: An Effective and Simple RNA-Seq Differential Gene Expression Pipeline Using Nextflow


Abstract
:1. Introduction
2. Materials and Methods
2.1. Implementation
2.1.1. Input
2.1.2. Preprocessing & Mapping
2.1.3. Quantification & Differential Gene Expression Analysis
2.1.4. Transcriptome Assembly
2.1.5. Output
2.2. DEG Validation of RNA-Seq with Publicly Available qRT-PCR Data
2.3. Transcriptome Assembly Validation
3. Results and Discussion
3.1. The Pipeline Produces Well-Structured Output and Comprehensive Insights into DEGs
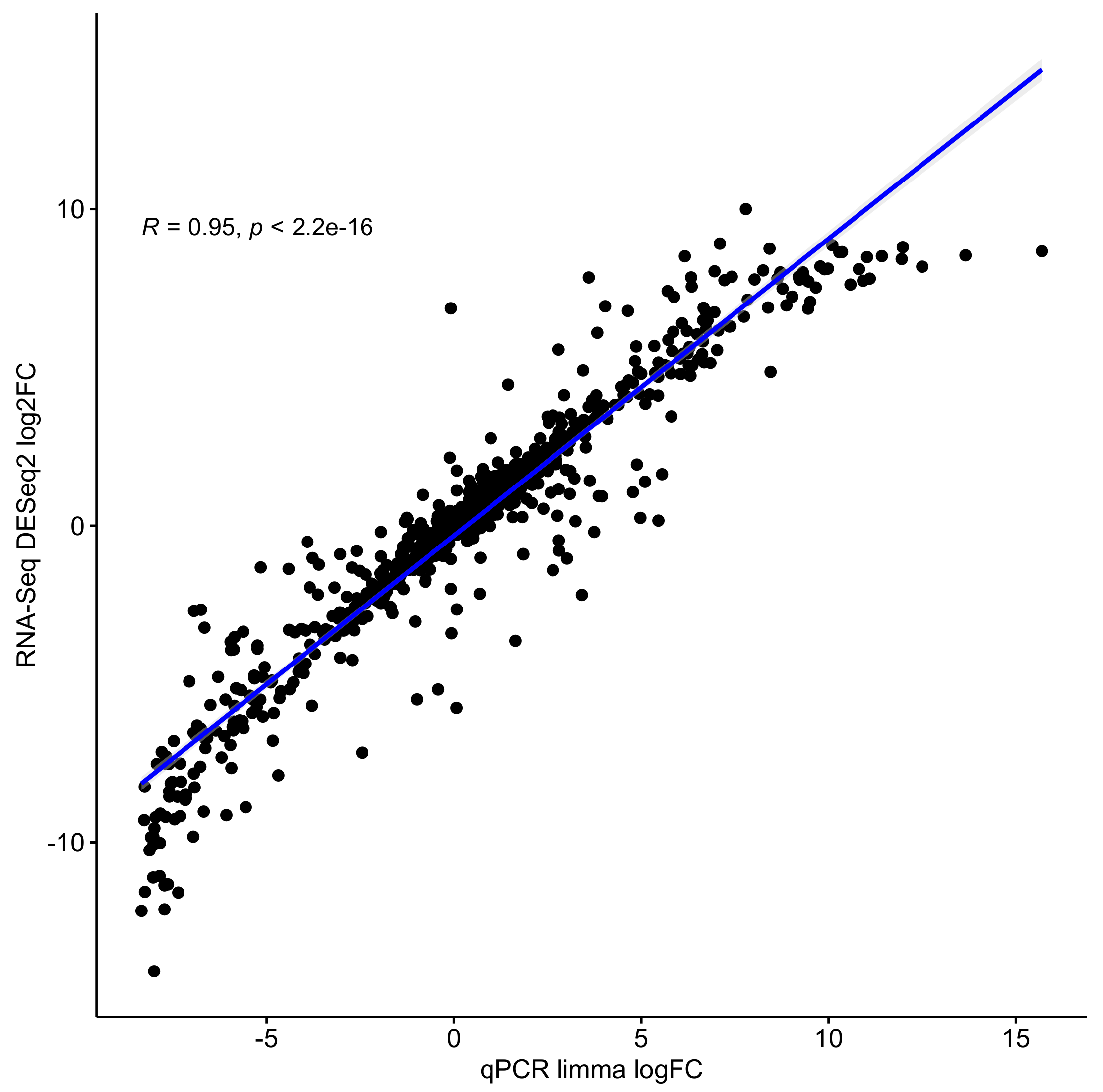
3.2. RNA-Seq Results of the Pipeline Correlate Strongly with qRT-PCR Results
3.3. Transcriptome Assembly Quality Is Comparable with Previously Reported Results
3.4. Comparison with Other RNA-Seq Pipelines
4. Conclusions
Future Direction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNAflow | VIPER [4] | Nextpresso [5] | TRAPLINE [6] | RNAsik [7] | hppRNA [8] | nf-core/rnaseq [9] | RCP [10] | RASflow [50] | OneStopRNAseq [53] | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pipeline scope | Read QC | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Trimming | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | |
| rRNA removal | ✓ | ✱ no automatic removal | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | |
| DEG analysis | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Pathway & downstream analysis | ✱ for some species | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✱ | |
| Comprehensive reports | ✓ | ✓ | ✗ | ✗ | ✱ | ✗ | ✓ | ✗ | ✓ | ✓ | |
| Transcriptomics extension | ✱ only assembly | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | |
| Implementation | Workflow management/ platform | Nextflow | Snakemake | RUbioSeq | Galaxy | BigDataScript | Partly Snakemake | Nextflow | ✗ | Snakemake | Snakemake (backend) |
| HPC support | via Nextflow | via Snakemake | SGE, PBS | via Galaxy | via BDS | ✗ | via Nextflow | Slurm | via Snakemake | via Snakemake | |
| Minimal input | ✓ | ✱ | ✗ | ✓ | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | |
| Easy accessible documentation | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | |
| Reproducibility | Build in revision specific execution | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ |
| Containerization/ dependency management | Conda, Docker, Singularity | Conda (no encapsulation) | Docker (no encapsulation) | via Galaxy | Conda (no encapsulation) | ✗ | Conda, Docker, Singularity, Podman | ✗ | Conda | Conda, Singularity |
References
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiss, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermüller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, J.; Dumontier, S.; Gosselin, R.; Scott, M.S. Current RNA-seq methodology reporting limits reproducibility. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornwell, M.; Vangala, M.; Taing, L.; Herbert, Z.; Köster, J.; Li, B.; Sun, H.; Li, T.; Zhang, J.; Qiu, X.; et al. VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinform. 2018, 19, 135. [Google Scholar] [CrossRef] [PubMed]
- Graña, O.; Rubio-Camarillo, M.; Fdez-Riverola, F.; Pisano, D.; Glez-Peña, D. Nextpresso: Next generation sequencing expression analysis pipeline. Curr. Bioinform. 2018, 13, 583–591. [Google Scholar] [CrossRef]
- Wolfien, M.; Rimmbach, C.; Schmitz, U.; Jung, J.J.; Krebs, S.; Steinhoff, G.; David, R.; Wolkenhauer, O. TRAPLINE: A standardized and automated pipeline for RNA sequencing data analysis, evaluation and annotation. BMC Bioinform. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tsyganov, K.; Perry, A.J.; Archer, S.K.; Powell, D. RNAsik: A Pipeline for complete and reproducible RNA-seq analysis that runs anywhere with speed and ease. J. Open Source Softw. 2018, 3, 583. [Google Scholar] [CrossRef]
- Wang, D. hppRNA—A Snakemake-based handy parameter-free pipeline for RNA-Seq analysis of numerous samples. Briefings Bioinform. 2018, 19, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Hammarén, R.; Peltzer, A.; Moreno, D.; Garcia, M.; rfenouil; marchoeppner; Panneerselvam, S.; Sven, F.; jun-wan; et al. nf-core/rnaseq: Nf-core/rnaseq version 1.4.2. 2019. Available online: https://zenodo.org/record/3503887#.X9Hk0LMRVPY (accessed on 9 December 2020). [CrossRef]
- Overbey, E.G.; Saravia-Butler, A.M.; Zhang, Z.; Rathi, K.S.; Fogle, H.; da Silveira, W.A.; Barker, R.J.; Bass, J.J.; Beheshti, A.; Berrios, D.C.; et al. NASA GeneLab RNA-Seq Consensus Pipeline: Standardized Processing of Short-Read RNA-Seq Data. bioRxiv 2020. Available online: https://www.biorxiv.org/content/early/2020/11/10/2020.11.06.371724.full.pdf (accessed on 9 December 2020).
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Merkel, D. Docker: Lightweight linux containers for consistent development and deployment. Linux J. 2014, 2014, 2. [Google Scholar]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Wallace, E.; Kavoussanakis, K. Using rapid prototyping to choose a bioinformatics workflow management system. bioRxiv 2020. Available online: https://www.biorxiv.org/content/early/2020/08/05/2020.08.04.236208.full.pdf (accessed on 9 December 2020).
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Mount, S.M.; Kingsford, C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014, 32, 462–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corchete, L.A.; Rojas, E.A.; Alonso-López, D.; De Las Rivas, J.; Gutiérrez, N.C.; Burguillo, F.J. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Harrington, C.A.; Fei, S.S.; Minnier, J.; Carbone, L.; Searles, R.; Davis, B.A.; Ogle, K.; Planck, S.R.; Rosenbaum, J.T.; Choi, D. RNA-Seq of human whole blood: Evaluation of globin RNA depletion on Ribo-Zero library method. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Sheth, R.U.; Kaufman, A.; Wang, H.H. Scalable and cost-effective ribonuclease-based rRNA depletion for transcriptomics. Nucleic Acids Res. 2019, 48, e20. Available online: https://academic.oup.com/nar/article-pdf/48/4/e20/32615496/gkz1169.pdf (accessed on 9 December 2020). [CrossRef] [Green Version]
- Fauver, J.R.; Akter, S.; Morales, A.I.O.; Black, W.C.; Rodriguez, A.D.; Stenglein, M.D.; Ebel, G.D.; Weger-Lucarelli, J. A reverse-transcription/RNase H based protocol for depletion of mosquito ribosomal RNA facilitates viral intrahost evolution analysis, transcriptomics and pathogen discovery. Virology 2019, 528, 181–197. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Simoneau, J.; Gosselin, R.; Scott, M.S. Factorial study of the RNA-seq computational workflow identifies biases as technical gene signatures. NAR Genom. Bioinform. 2020, 2, 2. [Google Scholar] [CrossRef]
- Schaarschmidt, S.; Fischer, A.; Zuther, E.; Hincha, D.K. Evaluation of Seven Different RNA-Seq Alignment Tools Based on Experimental Data from the Model Plant Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 1720. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. Available online: https://cancerres.aacrjournals.org/content/77/21/e31.full.pdf (accessed on 9 December 2020). [CrossRef] [Green Version]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Huntley, M.A.; Larson, J.L.; Chaivorapol, C.; Becker, G.; Lawrence, M.; Hackney, J.A.; Kaminker, J.S. ReportingTools: An automated result processing and presentation toolkit for high-throughput genomic analyses. Bioinformatics 2013, 29, 3220–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado-Torres, L.; Jaffe, A.E.; Leek, J.T. regionReport: Interactive reports for region-level and feature-level genomic analyses. F1000Research 2015, 4, 105. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Hölzer, M.; Marz, M. De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-Seq assemblers. Gigascience 2019, 8, giz039. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [Green Version]
- MAQC Consortium; Shi, L.; Reid, L.H.; Jones, W.D.; Shippy, R.; Warrington, J.A.; Baker, S.C.; Collins, P.J.; de Longueville, F.; Kawasaki, E.S.; et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006, 24, 1151–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canales, R.D.; Luo, Y.; Willey, J.C.; Austermiller, B.; Barbacioru, C.C.; Boysen, C.; Hunkapiller, K.; Jensen, R.V.; Knight, C.R.; Lee, K.Y.; et al. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat. Biotechnol. 2006, 24, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Liu, J.; Li, G.; Chang, Z.; Yu, T.; Liu, B.; McMullen, R.; Chen, P.; Huang, X. BinPacker: Packing-based de novo transcriptome assembly from RNA-seq data. PLoS Comput. Biol. 2016, 12, e1004772. [Google Scholar] [CrossRef] [PubMed]
- MacManes, M.D. The Oyster River Protocol: A multi-assembler and kmer approach for de novo transcriptome assembly. PeerJ 2018, 6, e5428. [Google Scholar] [CrossRef]
- Freedman, A.H.; Clamp, M.; Sackton, T.B. Error, noise and bias in de novo transcriptome assemblies. Mol. Ecol. Resour. 2020, 1–12. [Google Scholar] [CrossRef]
- Hölzer, M. A decade of de novo transcriptome assembly: Are we there yet? Mol. Ecol. Resour. 2020, 1–3. [Google Scholar] [CrossRef]
- Zhang, X.; Jonassen, I. RASflow: An RNA-Seq analysis workflow with Snakemake. BMC Bioinform. 2020, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Köster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. Available online: https://academic.oup.com/bioinformatics/article-pdf/28/19/2520/819790/bts480.pdf (accessed on 9 December 2020). [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. Available online: https://academic.oup.com/nar/article-pdf/46/W1/W537/25110642/gky379.pdf (accessed on 9 December 2020). [CrossRef] [Green Version]
- Li, R.; Hu, K.; Liu, H.; Green, M.R.; Zhu, L.J. OneStopRNAseq: A Web Application for Comprehensive and Efficient Analyses of RNA-Seq Data. Genes 2020, 11, 1165. [Google Scholar] [CrossRef] [PubMed]
- Perkel, J.M. Workflow systems turn raw data into scientific knowledge. Natur 2019, 573, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarantopoulou, D.; Tang, S.Y.; Ricciotti, E.; Lahens, N.F.; Lekkas, D.; Schug, J.; Guo, X.S.; Paschos, G.K.; FitzGerald, G.A.; Pack, A.I.; et al. Comparative evaluation of RNA-Seq library preparation methods for strand-specificity and low input. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzberg, S.L. Next-generation genome annotation: We still struggle to get it right. Genome Biol 2019, 20, 92. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lataretu, M.; Hölzer, M. RNAflow: An Effective and Simple RNA-Seq Differential Gene Expression Pipeline Using Nextflow. Genes 2020, 11, 1487. https://doi.org/10.3390/genes11121487
Lataretu M, Hölzer M. RNAflow: An Effective and Simple RNA-Seq Differential Gene Expression Pipeline Using Nextflow. Genes. 2020; 11(12):1487. https://doi.org/10.3390/genes11121487
Chicago/Turabian StyleLataretu, Marie, and Martin Hölzer. 2020. "RNAflow: An Effective and Simple RNA-Seq Differential Gene Expression Pipeline Using Nextflow" Genes 11, no. 12: 1487. https://doi.org/10.3390/genes11121487
APA StyleLataretu, M., & Hölzer, M. (2020). RNAflow: An Effective and Simple RNA-Seq Differential Gene Expression Pipeline Using Nextflow. Genes, 11(12), 1487. https://doi.org/10.3390/genes11121487