Genome-Wide Association Study (GWAS) for Resistance to Sclerotinia sclerotiorum in Common Bean


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. White Mold Response Evaluation
2.3. Data Analysis
2.4. Genome-Wide Association Study
2.5. Quantitative Trait Intervals
2.6. Candidate Gene Identification
3. Results
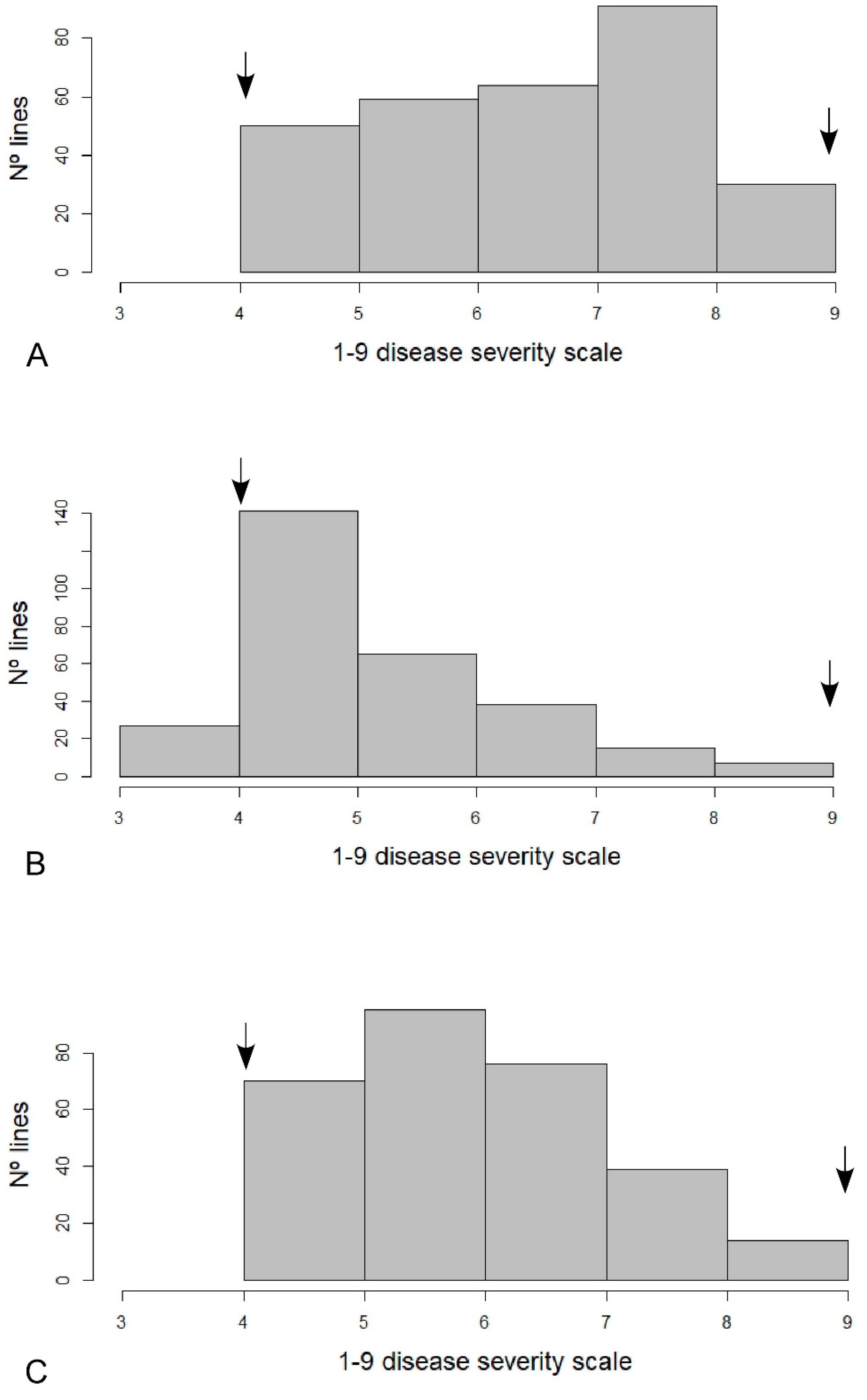
3.1. White Mold Evaluation
3.2. Genome-Wide Association Study
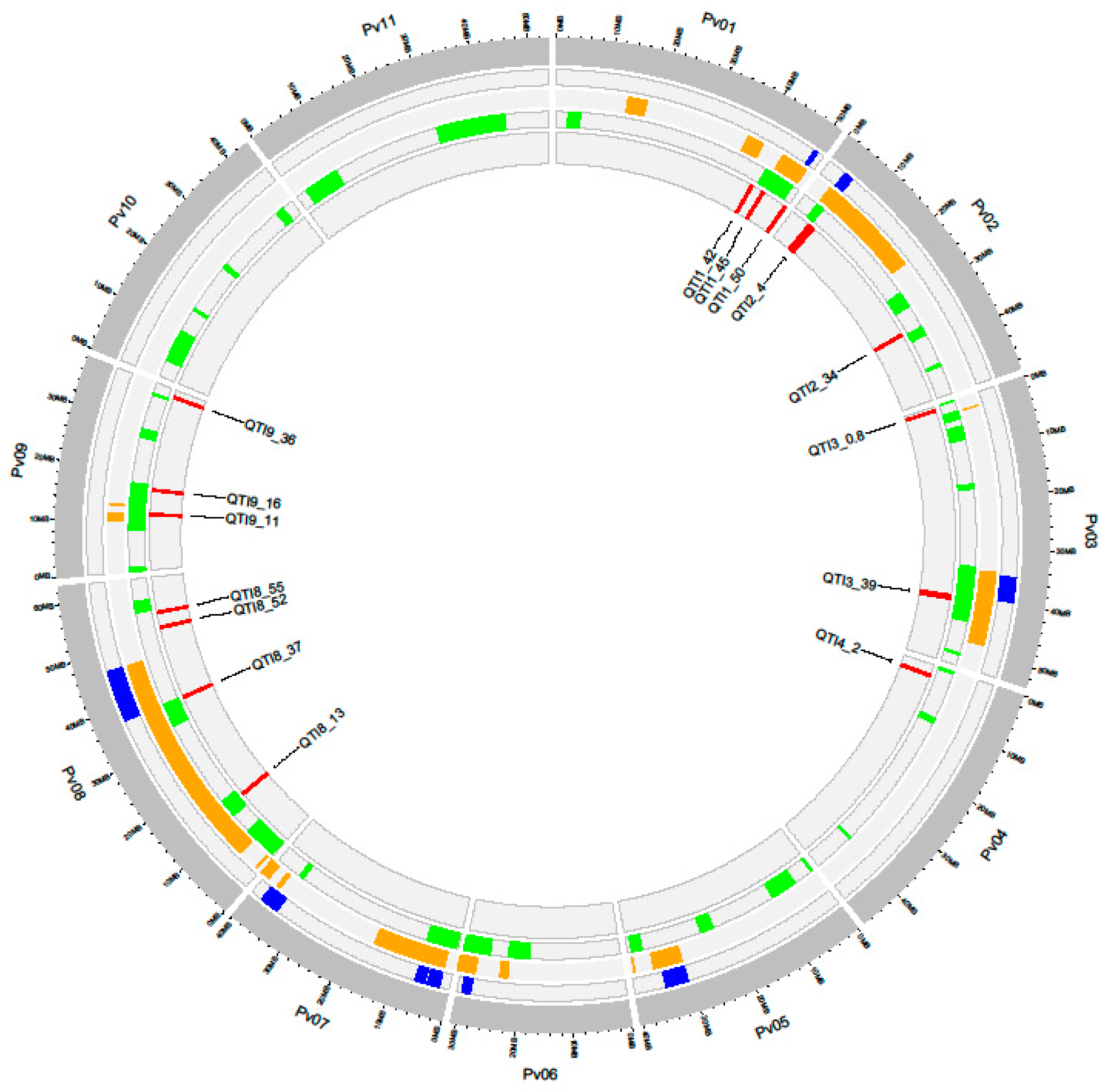
3.3. Quantitative Trait Intervals
3.4. Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. FAOSTAT Statistics Database; FAO: Rome, Italy, 1998. [Google Scholar]
- Gepts, P.; Osborne, T.C.; Rashka, K.; Bliss, F.A. Phaseolin protein variability in wild forms and landraces of the common bean (Phaseolus vulgaris L.): Evidence for multiple centers of domestication. Econ. Bot. 1986, 40, 451–468. [Google Scholar] [CrossRef]
- Koenig, R.L.; Gepts, P. Allozyme diversity in wild Phaseolus vulgaris: Further evidence for two major centers of genetic diversity. Theor. Appl. Genet. 1989, 78, 809–817. [Google Scholar] [CrossRef]
- Singh, S.P.; Gepts, P.; Debouck, D.G. Races of common bean (Phaseolus vulgaris, Fabaceae). Econ. Bot. 1991, 45, 379–396. [Google Scholar] [CrossRef]
- Kwak, M.; Gepts, P. Structure of genetic diversity in the two major gene pools of common bean (Phaseolus vulgaris L., Fabaceae). Theor. Appl. Genet. 2009, 118, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Bitocchi, E.; Bellucci, E.; Nanni, L.; Rau, D.; Attene, G.; Papa, R. Linkage disequilibrium and population structure in wild and domesticated populations of Phaseolus vulgaris L. Evol. Appl. 2009, 2, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Campa, A.; Murube, E.; Ferreira, J.J. Genetic diversity, population structure, and linkage disequilibrium in a Spanish common bean diversity panel revealed through genotyping-by-sequencing. Genes 2018, 9, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolland, G.J.; Hall, R. Index of plant host of Sclerotinia sclerotiorum. Can. J. Plant Pathol. 1994, 16, 93–108. [Google Scholar] [CrossRef]
- Schwartz, H.F.; Steadman, J.R.; Hall, R.; Forster, R.L. Compendium of Bean Diseases, 2nd ed.; APS Press: Fort Collins, CO, USA, 2005; p. 109. ISBN 0890543283. [Google Scholar]
- Kabbage, M.; Yarden, O.; Dickman, M.B. Pathogenic attributes of Sclerotinia sclerotiorum: Switching from a biotrophic to necrotrophic lifestyle. Plant Sci. 2015, 233, 53–60. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, L.-Y.; Cao, J.; Li, Y.-L.; Ding, L.-N.; Zhu, K.-M.; Yang, Y.-H.; Tan, X.-L. Recent advances in mechanism of plant defense to Sclerotinia sclerotiorum. Front. Plant Sci. 2019, 10, 1314. [Google Scholar] [CrossRef] [PubMed]
- Miklas, P.N.; Delorme, R.; Johnson, W.C.; Gepts, P. QTL conditioning physiological resistance and avoidance to white mold in dry bean. Crop Sci. 2001, 41, 309–315. [Google Scholar] [CrossRef]
- Park, O.S.; Coyne, D.P.; Steadman, J.R.; Skroch, P.W. Mapping of QTL for resistance to white mold disease in common bean. Crop Sci. 2001, 41, 1253–1262. [Google Scholar] [CrossRef]
- Kolkman, J.M.; Kelly, J.D. QTL conferring resistance and avoidance to white mold in common bean. Crop Sci. 2003, 43, 539–548. [Google Scholar] [CrossRef]
- Miklas, P.N.; Delorme, R. Identification of QTL conditioning resistance to white mold in snap bean. J. Am. Soc. Hortic. Sci. 2003, 128, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Ender, M.; Kelly, J.D. Identification of QTL associated with white mold resistance in common bean. Crop Sci. 2005, 45, 2482–2490. [Google Scholar] [CrossRef]
- Maxwell, J.J.; Brick, M.A.; Byrne, P.F.; Schwartz, H.F.; Shan, X.; Ogg, J.B.; Hensen, R.A. Quantitative trait loci linked to white mold resistance in common bean. Crop Sci. 2007, 47, 2285–2294. [Google Scholar] [CrossRef]
- Miklas, P.N.; Larsen, K.M.; Terpstra, K.A.; Hauf, D.C.; Grafton, K.F.; Kelly, J.D. QTL analysis of ICA Bunsi-derived resistance to white mold in a Pinto 3 navy bean cross. Crop Sci. 2007, 47, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Soule, M.; Porter, L.; Medina, J.; Santana, G.P.; Blair, M.W.; Miklas, P.N. Comparative QTL map for white mold resistance in common bean, and characterization of partial resistance in dry bean lines VA19 and I9365-31. Crop Sci. 2011, 51, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Vega, E.; Pascual, A.; Campa, A.; Giraldez, R.; Miklas, P.N.; Ferreira, J.J. Mapping quantitative trait loci conferring partial physiological resistance to white mold in the common bean RIL population Xana x Cornell49242. Mol. Breed. 2012, 29, 31–41. [Google Scholar] [CrossRef]
- Miklas, P.N.; Porter, L.D.; Kelly, J.D.; Myers, J.R. Characterization of white mold disease avoidance in common bean. Eur. J. Plant Pathol. 2013, 135, 525–543. [Google Scholar] [CrossRef] [Green Version]
- Hoyos-Villegas, V.; Mkwaila, W.; Cregan, P.B.; Kelly, J.D. Quantitative trait loci analysis of white mold avoidance in pinto bean. Crop Sci. 2015, 55, 2116–2129. [Google Scholar] [CrossRef]
- Vasconcellos, R.C.C.; Oraguzie, O.B.; Soler, A.; Arkwazee, H.; Myers, J.R.; Ferreira, J.J.; Song, Q.; McClean, P.; Miklas, P.N. Meta-QTL for resistance to white mold in common bean. PLoS ONE 2017, 12, e0171685. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.M.; Mao, Y.; Xie, C.; Smith, H.; Luo, L.; Xu, S. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics 2005, 169, 2267–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Feng, J.Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. pLARmEB: Integration of least angle regression with empirical bayes for multi-locus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Tamba, C.L.; Ni, Y.L.; Zhang, Y.M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multilocus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Tamba, C.L. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018. [Google Scholar] [CrossRef]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.L.; Wen, Y.J.; Dunwell, J.M.; Zhang, Y.M. pKWmEB: Integration of Kruskal-Wallis test with empirical bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Arkwazee, H.; Hart, J.; Porch, T.; Griffiths, P.; Davis, J.; Myers, J.R. Genome wide association study (GWAS) for white mold resistance in snap bean. Annu. Rept. Bean Improv. Coop. 2018, 61, 85. [Google Scholar]
- Pérez-Vega, E.; Campa, A.; De la Rosa, L.; Giraldez, R.; Ferreira, J.J. Genetic diversity in a core collection established from the main genebank in Spain. Crop Sci. 2009, 49, 1377–1386. [Google Scholar] [CrossRef]
- Pascual, A.; Campa, A.; Pérez-Vega, E.; Giraldez, R.; Miklas, P.N.; Ferreira, J.J. Screening common bean for resistance to four Sclerotinia sclerotiorum isolates collected in northern Spain. Plant Dis. 2010, 94, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Petzoldt, R.; Dickson, M.H. Straw test for resistance to white mold in beans. Annu. Rep. Bean Improv. Coop. Meet 1996, 39, 142–143. [Google Scholar]
- Miklas, P.N. Marker-assisted backcrossing QTL for partial resistance to Sclerotinia white mold in dry bean. Crop Sci. 2007, 47, 935–942. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; ISBN 3-900051-07-0. Available online: http://www.R-project.org (accessed on 1 April 2020).
- Zhang, Y.W.; Tamba, C.L.; Wen, Y.J.; Li, P.; Ren, W.L.; Ni, Y.L.; Gao, J.; Zhang, Y.M. mrMLM v4.0: An R platform for multi-locus genome-wide association studies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Moghaddam, S.M.; Mamidi, S.; Osorno, J.M.; Lee, R.; Brick, M.; Kelly, J.; Miklas, P.; Urrea, C.; Song, Q.; Cregan, P.; et al. Genome-Wide association study identifies candidate loci underlying agronomic traits in a middle american diversity panel of common bean. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ouyang, Y.; Yao, W. ShinyCircos: An R/shiny application for interactive creation of circos plot. Bioinformatics 2018, 34, 1229–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, P.A.; Coyne, D.P.; Steadman, J.R. Inheritance of resistance to white mold disease in a diallel cross of dry beans. Crop Sci. 1984, 24, 929–933. [Google Scholar] [CrossRef]
- Miklas, P.N.; Hauf, D.C.; Henson, R.A.; Grafton, K.F. Inheritance of ICA Bunsi derived resistance in a navy × pinto bean cross. Crop Sci. 2004, 44, 1584–1588. [Google Scholar] [CrossRef] [Green Version]
- Meziadi, C.; Richard, M.M.S.; Derquennes, A.; Thareau, V.; Blanchet, S.; Gratias, A.; Pflieger, S.; Geffroy, V. Development of molecular markers linked to disease resistance genes in common bean based on whole genome sequence. Plant Sci. 2016, 242, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Campa, A.; Ferreira, J.J. Gene coding for an elongation factor is involved in resistant against powdery mildew in common bean. Theor. Appl. Genet. 2017, 130, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Murube, E.; Campa, A.; Ferreira, J.J. Integrating genetic and physical positions of the anthracnose resistance genes described in bean chromosomes Pv01 and Pv04. PLoS ONE 2019, 14, e0212298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.; Velasco, D.; Gepts, P. Mapping homologous sequences for determinacy and photoperiod sensitivity in common bean (Phaseolus vulgaris). J. Hered. 2008, 99, 283–291. [Google Scholar] [CrossRef]
- Repinski, S.L.; Kwak, M.; Gepts, P. The common bean growth habit gene PvTFL1y is a functional homolog of Arabidopsis TFL1. Theor. Appl. Genet. 2012, 124, 1539–1547. [Google Scholar] [CrossRef]
- Pandian, B.A.; Sathishraj, R.; Djanaguiraman, M.; Prasad, P.V.V.; Jugulam, M. Role of cytochrome P450 enzymes in plant stress response. Antioxidants 2020, 9, 454. [Google Scholar] [CrossRef]
- Yan, Q.; Cui, X.; Lin, S.; Gan, S.; Xing, H.; Dou, D. GmCYP82A3, a soybean cytochrome P450 family gene involved in the jasmonic acid and ethylene signaling pathway, enhances plant resistance to biotic and abiotic stresses. PLoS ONE 2016, 11, e0162253. [Google Scholar] [CrossRef] [Green Version]
- Sessa, G.; Martin, G.B. Protein kinases in the plant defence response. Adv. Bot. Res. 2000, 32, 379–404. [Google Scholar] [CrossRef]
- Marone, D.; Russo, M.A.; Laido, G.; Leonardis, A.M.; Mastrangelo, A.M. Plant nucleotide binding site-leucine-rich repeat (NBS-LRR) genes: Active guardians in host defense response. Int J. Mol. Sci. 2013, 14, 7302–7326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Stotz, H.U. Defense against Sclerotinia sclerotiorum in arabidopsis is dependent on jasmonic acid, salicylic acid, and ethylene signaling xiaomei. Mol. Plant Microbe. Interact. 2007, 20, 1384–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Wang, J.; An, L.; Doerge, R.W.; Chen, Z.J.; Grau, C.R.; Meng, J.; Osborn, T.C. Analysis of gene expression profiles in response to Sclerotinia sclerotiorum in Brassica napus. Planta 2007, 227, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Kalunke, R.M.; Janni, M.; Sella, L.; David, P.; Geffroy, V.; Favaron, F.; D´Ovidio, R. Transcript analysis of the bean polygalacturonase-inhibiting protein gene family reveals that Pvpgip2 is expressed in the whole plant and is strongly induced by pathogen infection. J. Plant Pathol. 2011, 93, 141–148. [Google Scholar] [CrossRef]
- D´Ovidio, R.; Raiola, A.; Capodicasa, C.; Devoto, A.; Pontiggia, D.; Roberti, S.; Galletti, R.; Conti, E.; O’Sullivan, D.; De Lorenzo, G. Characterization of the complex locus of bean encoding Polygalacturonase-Inhibiting Proteins reveals subfunctionalization for defense against fungi and insects. Plant Physiol. 2004, 135, 2424–2435. [Google Scholar] [CrossRef] [Green Version]
- Vasconcellos, R.C.C.; Lima, T.F.C.; Fernandes-Brum, C.N.; Chalfun-Junior, A.; Santos, J.B. Expression and validation of PvPGIP genes for resistance to white mold (Sclerotinia sclerotiorum) in common beans (Phaseolus vulgaris L.). Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Porto, A.C.M.; Cardon, C.H.; Vasconcellos, R.C.C.; Novaes, E.; Leite, M.E.; Chalfun-Junior, A.; Pereira, W.A.; Santos, J.B. Expression of candidate genes related to white mold resistance in common beans. Trop. Plant Pathol. 2019, 44, 483–493. [Google Scholar] [CrossRef]
- Visel, A.; Rubin, E.M.; Pennacchio, L.A. Genomic views of distant-acting enhancers. Nature 2009, 461, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How far from the SNP may the causative genes be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| SDP | Accession/Variety | Type of Material | GP | GH | E1 | E2 | Em |
|---|---|---|---|---|---|---|---|
| SDP194 | BGE043359 | Spanish landrace | A | Ind | 4.0 | 4.0 | 4.0 |
| SDP217 | CN_227 | Spanish landrace | M | Ind | 4.0 | 4.0 | 4.0 |
| SDP289 | Sacha | Commercial variety | MA | Ind | 4.2 | 4.0 | 4.1 |
| SDP203 | BILMA | Commercial variety | MA | Ind | 4.3 | 4.0 | 4.1 |
| SDP216 | CN_226 | Spanish landrace | M | Ind | 4.3 | 4.0 | 4.1 |
| SDP246 | Helda | Commercial variety | MA | Ind | 4.1 | 4.1 | 4.1 |
| SDP274 | PLANETA | Commercial variety | MA | Ind | 4.1 | 4.3 | 4.2 |
| SDP226 | DONNA | Commercial variety | MA | Ind | 4.3 | 4.2 | 4.2 |
| SDP108 | BGE011762 | Spanish landrace | A | Ind | 4.3 | 4.3 | 4.3 |
| SDP039 | BGE003139 | Spanish landrace | MA | Ind | 4.2 | 4.4 | 4.3 |
| SDP133 | BGE022831 | Spanish landrace | MA | Ind | 4.7 | 4.0 | 4.3 |
| SDP305 | VITALIS | Commercial variety | MA | Ind | 4.5 | 4.1 | 4.3 |
| SDP234 | FLORENCIA | Commercial variety | MA | Ind | 4.4 | 4.3 | 4.4 |
| SDP304 | V381 | Spanish landrace | MA | Ind | 4.2 | 4.5 | 4.4 |
| SDP106 | BGE011736 | Spanish landrace | M | Det | 4.8 | 4.0 | 4.4 |
| SDP107 | BGE011758 | Spanish landrace | MA | Ind | 4.6 | 3.7 | 4.4 |
| SDP255 | MARCONI | Commercial variety | MA | Ind | 4.6 | 4.2 | 4.4 |
| SDP189 | BGE039982 | Spanish landrace | M | Ind | 4.6 | 4.1 | 4.4 |
| SDP025 | BGE002196 | Spanish landrace | M | Ind | 4.4 | 4.5 | 4.5 |
| SDP035 | BGE003121 | Spanish landrace | M | Ind | 4.5 | 4.5 | 4.5 |
| SDP053 | BGE003482 | Spanish landrace | A | Ind | 4.4 | 4.6 | 4.5 |
| SDP071 | BGE004000 | Spanish landrace | A | Ind | 5.0 | 4.0 | 4.5 |
| SDP088 | BGE005475 | Spanish landrace | MA | Ind | 5.2 | 3.8 | 4.5 |
| SDP221 | CN_241 | Spanish landrace | A | Det | 4.6 | 4.4 | 4.5 |
| SDP295 | Tendergreen | Commercial variety | A | Det | 5.0 | 4.1 | 4.5 |
| SDP110 | BGE013962 | Spanish landrace | M | Ind | 4.9 | 4.0 | 4.5 |
| SDP150 | BGE025745 | Spanish landrace | A | Ind | 5.0 | 4.1 | 4.5 |
| SDP024 | BGE002189 | Spanish landrace | MA | Ind | 5.0 | 4.0 | 4.5 |
| AB136 | Resistant check | MA | Ind | 4.0 | 4.0 | 4.0 | |
| Cornell49242 | Susceptible check | MA | Ind | 8.9 | 9.0 | 9.0 |
| E1 | E2 | Em | |||
|---|---|---|---|---|---|
| QTN | SNP | GWAS Method | −log10(p) | −log10(p) | −log10(p) |
| WM1_42.55 | s1_42558186 | MLM | 3 | - | 3 |
| WM1_42.85 | s1_42857105 | MLM | 4 | - | 3 |
| pLARmEB | 7 | - | 6 | ||
| WM1_42.91 | s1_42916300 | FASTmrEMMA | 7 | - | 5 |
| WM1_45.45 | s1_45457672 | MLM | - | 4 | 3 |
| WM1_45.59 | s1_45594842 | MLM | - | 6 | 4 |
| mrMLM | - | 7 | 5 | ||
| FASTmrMLM | - | 9 | 10 | ||
| ISIS EM-BLASSO | - | 11 | 11 | ||
| FASTmrEMMA | - | 7 | 5 | ||
| pLARmEB | - | 6 | 8 | ||
| pLARmEB | 5 | - | 7 | ||
| WM1_51.06 | s1_51067156 | ISIS EM-BLASSO | 3 | - | 3 |
| WM2_4.62 | s2_4625990 | MLM | 9 | - | 5 |
| WM2_5.57 | s2_5576016 | pLARmEB | 3 | - | 3 |
| WM2_34.64 | s2_34644216 | MLM | - | 5 | 4 |
| WM2_34.73 | s2_34738605 | mrMLM | 3 | - | 3 |
| WM3_0.9 | s3_947347 | FASTmrMLM | - | 4 | 5 |
| WM3_39.95 | s3_39951398 | MLM | 7 | - | 7 |
| pLARmEB | 3 | - | 3 | ||
| WM3_40.53 | s3_40535470 | MLM | - | 6 | 5 |
| WM4_2.11 | s4_2115855 | ISIS EM-BLASSO | - | 4 | 3 |
| WM4_2.37 | s4_2371248 | MLM | - | 7 | 6 |
| pKWmEB | - | 4 | 8 | ||
| pLARmEB | 4 | 3 | 5 | ||
| WM8_13.31 | s8_13317583 | MLM | - | 3 | 3 |
| WM8_37.13 | s8_37139214 | MLM | 4 | - | 4 |
| WM8_52.39 | s8_52395252 | MLM | 7 | - | 7 |
| FASTmrMLM | 6 | 6 | 7 | ||
| ISIS EM-BLASSO | 10 | - | 10 | ||
| FASTmrEMMA | - | 5 | 5 | ||
| pKWmEB | 5 | - | 4 | ||
| pLARmEB | - | 7 | 6 | ||
| WM8_55.56 | s8_55569419 | pKWmEB | 3 | - | 3 |
| WM9_11.70 | s9_11707260 | MLM | - | 5 | 4 |
| WM9_16.78 | s9_16783907 | mrMLM | - | 8 | 8 |
| WM9_36.67 | s9_36677215 | pLARmEB | 3 | - | 3 |
| QTI | Chr | Start-End | QTNs | Nº Annotated Genes | Nº Candidate Genes | Stage of Resistance Response (Number of Genes) |
|---|---|---|---|---|---|---|
| QTI1_42 | Pv01 | 42.46−43.02 | WM1_42.55, WM11_42.85, WM1_42.91 | 46 | 8 | Recognition (6), Signaling (2) |
| QTI1_45 | Pv01 | 45.36−45.69 | WM1_45.45, WM1_45.59 | 36 | 4 | Recognition (1), Signaling (1), Defense (2) |
| QTI1_50 | Pv01 | 50.97–51.17 | WM1_51.06 | 28 | 2 | Recognition (1), Defense (1) |
| QTI2_4 | Pv02 | 4.53–5.68 | WM2_4.62, WM2_5.57 | 74 | 7 | Recognition (3), Signaling (2), Defense (2) |
| QTI2_34 | Pv02 | 34.54–34.84 | WM2_34.64, WM2_34.73 | 23 | - | - |
| QTI3_0.9 | Pv03 | 0.80–1.05 | WM3_0.9 | 29 | 6 | Recognition (1), Defense (5) |
| QTI3_39 | Pv03 | 39.85–40.64 | WM3_39.95, WM3_40.53 | 77 | 7 | Recognition (2), Signaling (1), Defense (4) |
| QTI4_2 | Pv04 | 2.02–2.47 | WM4_2.11, WM4_2.37 | 42 | 12 | Recognition (1), Defense (11) |
| QTI8_13 | Pv08 | 13.22–13.42 | WM8_13.31 | 6 | - | - |
| QTI8_37 | Pv08 | 37.04–37.24 | WM8_37.13 | 2 | - | - |
| QTI8_52 | Pv08 | 52.30–52.50 | WM8_52.39 | 24 | 5 | Recognition (4), Defense (1) |
| QTI8_55 | Pv08 | 55.47–55.67 | WM8_55.56 | 22 | 1 | Signaling (1) |
| QTI9_11 | Pv09 | 11.61–11.88 | WM9_11.70 | 24 | 2 | Recognition (2) |
| QTI9_16 | Pv09 | 16.68–16.88 | WM9_16.78 | 15 | 3 | Recognition (1), Defense (2) |
| QTI9_36 | Pv09 | 36.58–36.78 | WM9_36.67 | 20 | 4 | Signaling (1), Defense (3) |
| 468 | 61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campa, A.; García-Fernández, C.; Ferreira, J.J. Genome-Wide Association Study (GWAS) for Resistance to Sclerotinia sclerotiorum in Common Bean. Genes 2020, 11, 1496. https://doi.org/10.3390/genes11121496
Campa A, García-Fernández C, Ferreira JJ. Genome-Wide Association Study (GWAS) for Resistance to Sclerotinia sclerotiorum in Common Bean. Genes. 2020; 11(12):1496. https://doi.org/10.3390/genes11121496
Chicago/Turabian StyleCampa, Ana, Carmen García-Fernández, and Juan José Ferreira. 2020. "Genome-Wide Association Study (GWAS) for Resistance to Sclerotinia sclerotiorum in Common Bean" Genes 11, no. 12: 1496. https://doi.org/10.3390/genes11121496
APA StyleCampa, A., García-Fernández, C., & Ferreira, J. J. (2020). Genome-Wide Association Study (GWAS) for Resistance to Sclerotinia sclerotiorum in Common Bean. Genes, 11(12), 1496. https://doi.org/10.3390/genes11121496