Genetic and Clinical Findings in an Ethnically Diverse Cohort with Retinitis Pigmentosa Associated with Pathogenic Variants in CERKL

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Studies
2.2. CERKL Mutation Screening
3. Results
3.1. Clinical Analysis
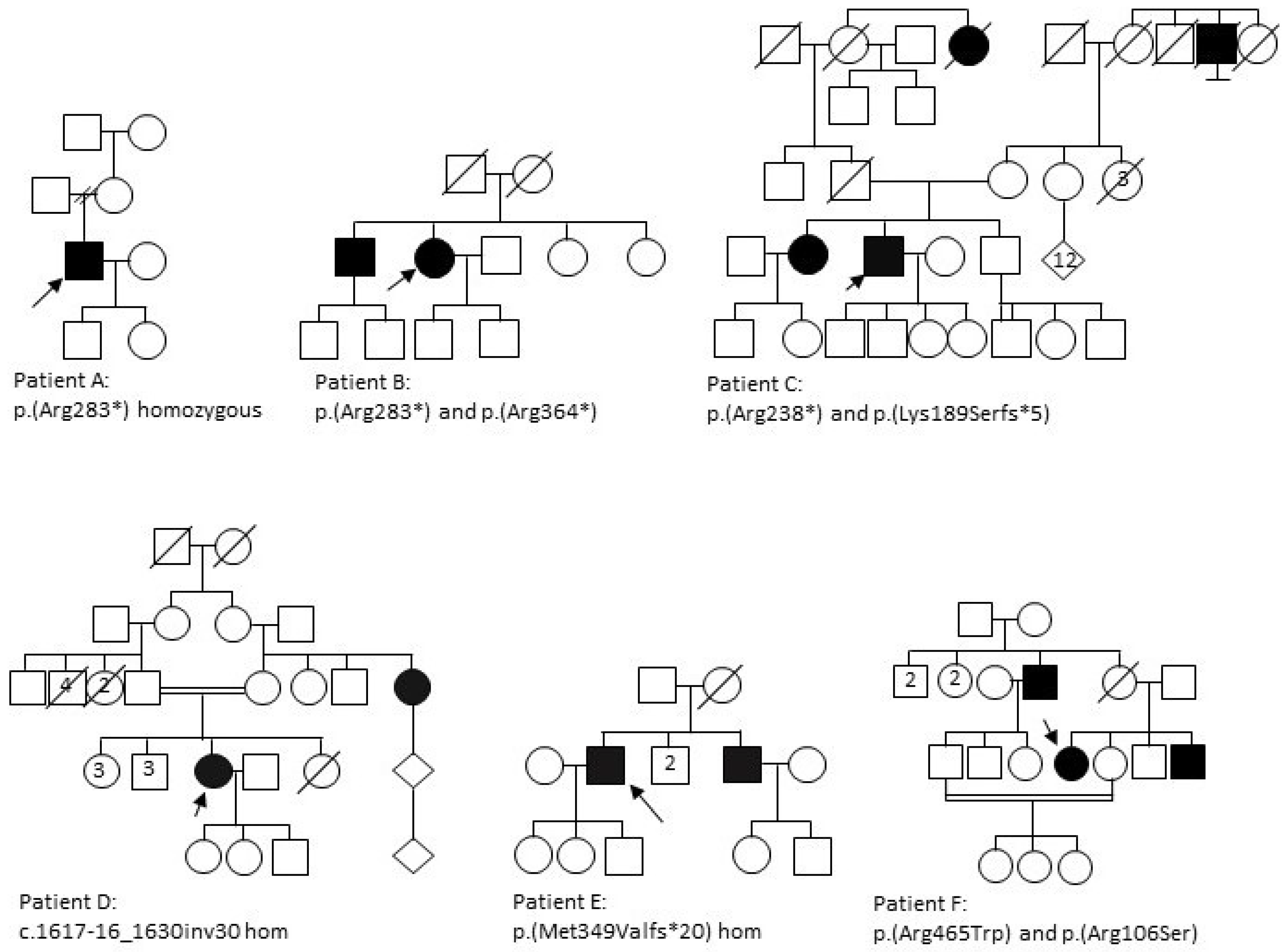
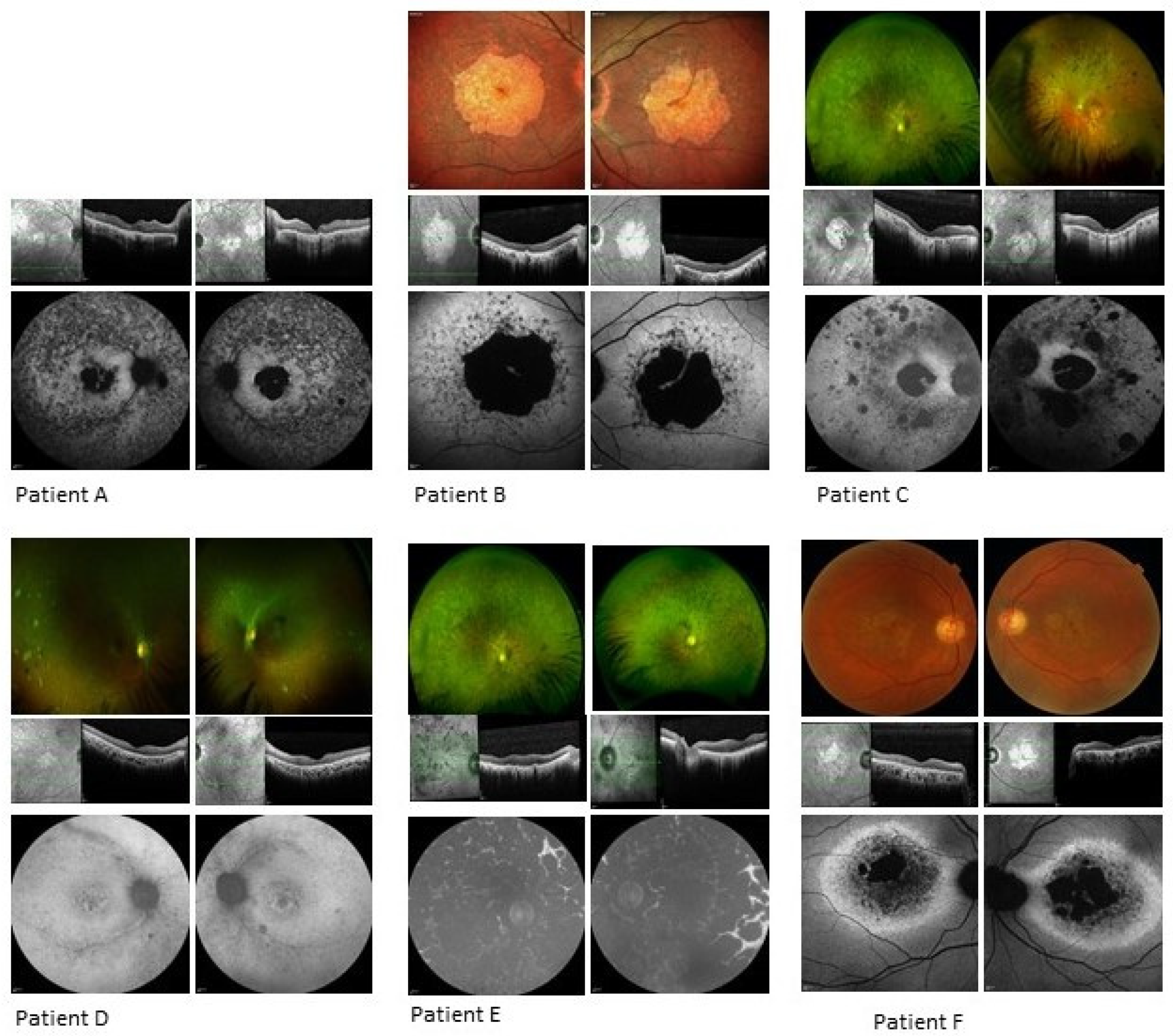
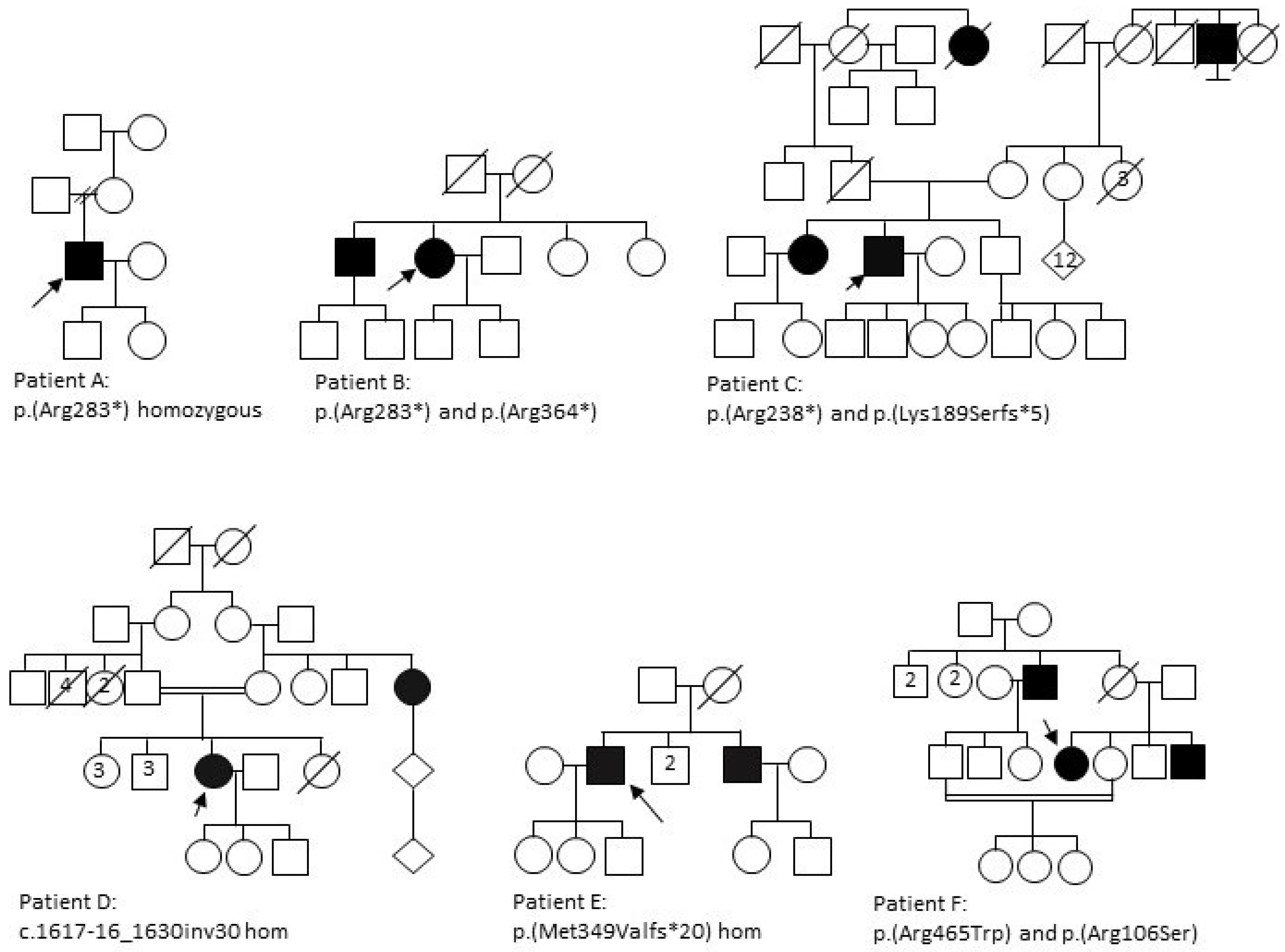
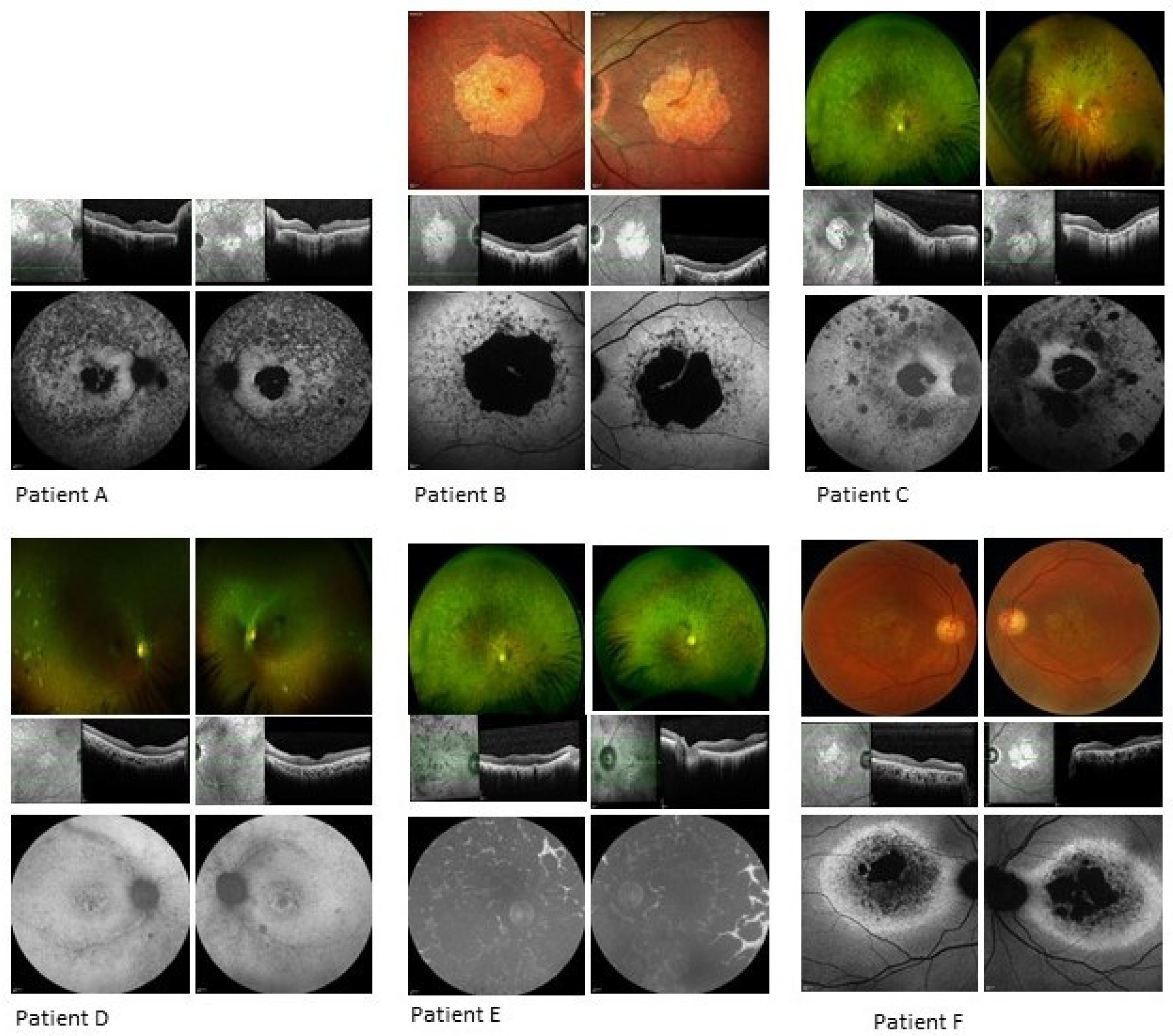
3.1.1. Patient A
3.1.2. Patient B
3.1.3. Patient C
3.1.4. Patient D
3.1.5. Patient E
3.1.6. Patient F
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Bayes, M.; Goldaracena, B.; Martinez-Mir, A.; Iragui-Madoz, M.I.; Solans, T.; Chivelet, P.; Bussaglia, E.; Ramos-Arroyo, M.A.; Baiget, M.; Vilageliu, L.; et al. A new autosomal recessive retinitis pigmentosa locus maps on chromosome 2q31-q33. J. Med. Genet. 1998, 35, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuson, M.; Marfany, G.; Gonzalez-Duarte, R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am. J. Hum. Genet. 2004, 74, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Munoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordonez, M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2016, 61, 51–62. [Google Scholar] [CrossRef]
- Bornancin, F.; Mechtcheriakova, D.; Stora, S.; Graf, C.; Wlachos, A.; Devay, P.; Urtz, N.; Baumruker, T.; Billich, A. Characterization of a ceramide kinase-like protein. Biochim. Biophys. Acta 2005, 1687, 31–43. [Google Scholar] [CrossRef]
- Domenech, E.B.; Andres, R.; Lopez-Iniesta, M.J.; Mirra, S.; Garcia-Arroyo, R.; Milla, S.; Sava, F.; Andilla, J.; Loza-Alvarez, P.; de la Villa, P.; et al. A New Cerkl Mouse Model Generated by CRISPR-Cas9 Shows Progressive Retinal Degeneration and Altered Morphological and Electrophysiological Phenotype. Investig. Ophthalmol. Vis. Sci. 2020, 61, 14. [Google Scholar] [CrossRef]
- Garanto, A.; Riera, M.; Pomares, E.; Permanyer, J.; de Castro-Miro, M.; Sava, F.; Abril, J.F.; Marfany, G.; Gonzalez-Duarte, R. High transcriptional complexity of the retinitis pigmentosa CERKL gene in human and mouse. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5202–5214. [Google Scholar] [CrossRef] [Green Version]
- Tuson, M.; Garanto, A.; Gonzalez-Duarte, R.; Marfany, G. Overexpression of CERKL, a gene responsible for retinitis pigmentosa in humans, protects cells from apoptosis induced by oxidative stress. Mol. Vis. 2009, 15, 168–180. [Google Scholar]
- Avela, K.; Sankila, E.M.; Seitsonen, S.; Kuuluvainen, L.; Barton, S.; Gillies, S.; Aittomaki, K. A founder mutation in CERKL is a major cause of retinal dystrophy in Finland. Acta Ophthalmol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Auslender, N.; Sharon, D.; Abbasi, A.H.; Garzozi, H.J.; Banin, E.; Ben-Yosef, T. A common founder mutation of CERKL underlies autosomal recessive retinal degeneration with early macular involvement among Yemenite Jews. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5431–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibi, I.; Chebil, A.; Falfoul, Y.; Allaman-Pillet, N.; Kort, F.; Schorderet, D.F.; El Matri, L. Identifying mutations in Tunisian families with retinal dystrophy. Sci. Rep. 2016, 6, 37455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila-Fernandez, A.; Riveiro-Alvarez, R.; Vallespin, E.; Wilke, R.; Tapias, I.; Cantalapiedra, D.; Aguirre-Lamban, J.; Gimenez, A.; Trujillo-Tiebas, M.J.; Ayuso, C. CERKL mutations and associated phenotypes in seven Spanish families with autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2709–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Shanks, M.; Packham, E.; Williams, J.; Haysmoore, J.; MacLaren, R.E.; Nemeth, A.H.; Clouston, P.; Downes, S.M. Next generation sequencing using phenotype-based panels for genetic testing in inherited retinal diseases. Ophthalmic Genet. 2020, 41, 331–337. [Google Scholar] [CrossRef]
- Tang, Z.; Wang, Z.; Wang, Z.; Ke, T.; Wang, Q.K.; Liu, M. Novel compound heterozygous mutations in CERKL cause autosomal recessive retinitis pigmentosa in a nonconsanguineous Chinese family. Arch. Ophthalmol. 2009, 127, 1077–1078. [Google Scholar] [CrossRef] [Green Version]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016. [Google Scholar] [CrossRef] [Green Version]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Muller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Ellingford, J.M.; Campbell, C.; Barton, S.; Bhaskar, S.; Gupta, S.; Taylor, R.L.; Sergouniotis, P.I.; Horn, B.; Lamb, J.A.; Michaelides, M.; et al. Validation of copy number variation analysis for next-generation sequencing diagnostics. Eur. J. Hum. Genet. 2017, 25, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Ramprasad, V.L.; Soumittra, N.; Mohamed, M.D.; Jafri, H.; Rashid, Y.; Danciger, M.; McKibbin, M.; Kumaramanickavel, G.; Inglehearn, C.F. A missense mutation in the nuclear localization signal sequence of CERKL (p.R106S) causes autosomal recessive retinal degeneration. Mol. Vis. 2008, 14, 1960–1964. [Google Scholar] [PubMed]
- Weisschuh, N.; Mayer, A.K.; Strom, T.M.; Kohl, S.; Glockle, N.; Schubach, M.; Andreasson, S.; Bernd, A.; Birch, D.G.; Hamel, C.P.; et al. Mutation Detection in Patients with Retinal Dystrophies Using Targeted Next Generation Sequencing. PLoS ONE 2016, 11, e0145951. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.; Ratnapriya, R.; du Plessis, M.; Chaitankar, V.; Ramesar, R.S.; Swaroop, A. Molecular Diagnosis of Inherited Retinal Diseases in Indigenous African Populations by Whole-Exome Sequencing. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6374–6381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littink, K.W.; Koenekoop, R.K.; van den Born, L.I.; Collin, R.W.; Moruz, L.; Veltman, J.A.; Roosing, S.; Zonneveld, M.N.; Omar, A.; Darvish, M.; et al. Homozygosity mapping in patients with cone-rod dystrophy: Novel mutations and clinical characterizations. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5943–5951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Tearle, R.G.; Liu, Y.P.; Oh, E.C.; Miyake, N.; Benaglio, P.; Harper, S.; Koskiniemi-Kuendig, H.; Venturini, G.; Sharon, D.; et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc. Natl. Acad. Sci. USA 2013, 110, 16139–16144. [Google Scholar] [CrossRef] [Green Version]
- Azab, B.; Barham, R.; Ali, D.; Dardas, Z.; Rashdan, L.; Bijawi, M.; Maswadi, R.; Awidi, A.; Jafar, H.; Abu-Ameerh, M.; et al. Novel CERKL variant in consanguineous Jordanian pedigrees with inherited retinal dystrophies. Can. J. Ophthalmol. 2019, 54, 51–59. [Google Scholar] [CrossRef]
- Huang, X.F.; Huang, F.; Wu, K.C.; Wu, J.; Chen, J.; Pang, C.P.; Lu, F.; Qu, J.; Jin, Z.B. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Glockle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, V.W.; Feng, Y.; Tian, X.; Li, F.Y.; Truong, C.; Wang, G.; Chiang, P.W.; Lewis, R.A.; Wong, L.J. Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6213–6223. [Google Scholar] [CrossRef]
- de Castro-Miro, M.; Tonda, R.; Escudero-Ferruz, P.; Andres, R.; Mayor-Lorenzo, A.; Castro, J.; Ciccioli, M.; Hidalgo, D.A.; Rodriguez-Ezcurra, J.J.; Farrando, J.; et al. Novel Candidate Genes and a Wide Spectrum of Structural and Point Mutations Responsible for Inherited Retinal Dystrophies Revealed by Exome Sequencing. PLoS ONE 2016, 11, e0168966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Carro, R.; Corton, M.; Sanchez-Navarro, I.; Zurita, O.; Sanchez-Bolivar, N.; Sanchez-Alcudia, R.; Lelieveld, S.H.; Aller, E.; Lopez-Martinez, M.A.; Lopez-Molina, M.I.; et al. Panel-based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Sci. Rep. 2016, 6, 19531. [Google Scholar] [CrossRef] [PubMed]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppieters, F.; Van Schil, K.; Bauwens, M.; Verdin, H.; De Jaegher, A.; Syx, D.; Sante, T.; Lefever, S.; Abdelmoula, N.B.; Depasse, F.; et al. Identity-by-descent-guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet. Med. 2014, 16, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Pomares, E.; Marfany, G.; Brion, M.J.; Carracedo, A.; Gonzalez-Duarte, R. Novel high-throughput SNP genotyping cosegregation analysis for genetic diagnosis of autosomal recessive retinitis pigmentosa and Leber congenital amaurosis. Hum. Mutat. 2007, 28, 511–516. [Google Scholar] [CrossRef]
- Aleman, T.S.; Soumittra, N.; Cideciyan, A.V.; Sumaroka, A.M.; Ramprasad, V.L.; Herrera, W.; Windsor, E.A.; Schwartz, S.B.; Russell, R.C.; Roman, A.J.; et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5944–5954. [Google Scholar] [CrossRef] [Green Version]
- Clark, G.R.; Crowe, P.; Muszynska, D.; O’Prey, D.; O’Neill, J.; Alexander, S.; Willoughby, C.E.; McKay, G.J.; Silvestri, G.; Simpson, D.A. Development of a diagnostic genetic test for simplex and autosomal recessive retinitis pigmentosa. Ophthalmology 2010, 117, 2169–2177. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Cho, G.Y.; Paavo, M.; Lee, W.; White, E.; Jauregui, R.; Sparrow, J.R.; Allikmets, R.; Tsang, S.H. Hyperautofluorescent Dots are Characteristic in Ceramide Kinase Like-associated Retinal Degeneration. Sci. Rep. 2019, 9, 876. [Google Scholar] [CrossRef] [Green Version]
- Wolock, C.J.; Stong, N.; Ma, C.J.; Nagasaki, T.; Lee, W.; Tsang, S.H.; Kamalakaran, S.; Goldstein, D.B.; Allikmets, R. A case-control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genet. Med. 2019, 21, 2336–2344. [Google Scholar] [CrossRef]
- Nadeem, R.; Kabir, F.; Li, J.; Gradstein, L.; Jiao, X.; Rauf, B.; Naeem, M.A.; Assir, M.Z.; Riazuddin, S.; Ayyagari, R.; et al. Mutations in CERKL and RP1 cause retinitis pigmentosa in Pakistani families. Hum. Genome Var. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Munoz, A.; Aller, E.; Jaijo, T.; Gonzalez-Garcia, E.; Cabrera-Peset, A.; Gallego-Pinazo, R.; Udaondo, P.; Salom, D.; Garcia-Garcia, G.; Millan, J.M. Expanding the Clinical and Molecular Heterogeneity of Nonsyndromic Inherited Retinal Dystrophies. J. Mol. Diagn. 2020, 22, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Maitra, P.; Natarajan, S.; Sripriya, S.; Mathavan, S.; Bhende, M.; Manchegowda, P.T. CERKL mutation causing retinitis pigmentosa(RP) in Indian population—A genotype and phenotype correlation study. Ophthalmic Genet. 2020, 41, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Abu-Safieh, L. Rod-Cone Dystrophy with Initially Preserved Visual Acuity Despite Early Macular Involvement Suggests Recessive CERKL Mutations. Ophthalmic Genet. 2015, 36, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Duncan, J.L.; Maranhao, B.; Kozak, I.; Branham, K.; Gabriel, L.; Lin, J.H.; Barteselli, G.; Navani, M.; Suk, J.; et al. Genetic analysis of 10 pedigrees with inherited retinal degeneration by exome sequencing and phenotype-genotype association. Physiol. Genom. 2017, 49, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Boulanger-Scemama, E.; El Shamieh, S.; Demontant, V.; Condroyer, C.; Antonio, A.; Michiels, C.; Boyard, F.; Saraiva, J.P.; Letexier, M.; Souied, E.; et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: Mutation spectrum and new genotype-phenotype correlation. Orphanet J. Rare Dis. 2015, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Wang, F.; Wang, H.; Xu, F.; Zaneveld, J.E.; Ren, H.; Keser, V.; Lopez, I.; Tuan, H.F.; Salvo, J.S.; et al. Next-generation sequencing-based molecular diagnosis of a Chinese patient cohort with autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4158–4166. [Google Scholar] [CrossRef] [Green Version]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Consortium, W.G.S.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Friedman, J.S.; Sandberg, M.A.; Swaroop, A.; Berson, E.L.; Dryja, T.P. Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc. Natl. Acad. Sci. USA 2004, 101, 17819–17824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, N.M.; Valkenburg, D.; Lambertus, S.; Klevering, B.J.; Boon, C.J.F.; Holz, F.G.; Cremers, F.P.M.; Fleckenstein, M.; Hoyng, C.B.; Lindner, M.; et al. Foveal Sparing in Central Retinal Dystrophies. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3456–3467. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Li, C.; Biswas, L.; Hu, X.; Liu, F.; Reilly, J.; Liu, X.; Liu, Y.; Huang, Y.; Lu, Z.; et al. CERKL gene knockout disturbs photoreceptor outer segment phagocytosis and causes rod-cone dystrophy in zebrafish. Hum. Mol. Genet. 2017, 26, 2335–2345. [Google Scholar] [CrossRef]
- Li, C.; Wang, L.; Zhang, J.; Huang, M.; Wong, F.; Liu, X.; Liu, F.; Cui, X.; Yang, G.; Chen, J.; et al. CERKL interacts with mitochondrial TRX2 and protects retinal cells from oxidative stress-induced apoptosis. Biochim. Biophys. Acta 2014, 1842, 1121–1129. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
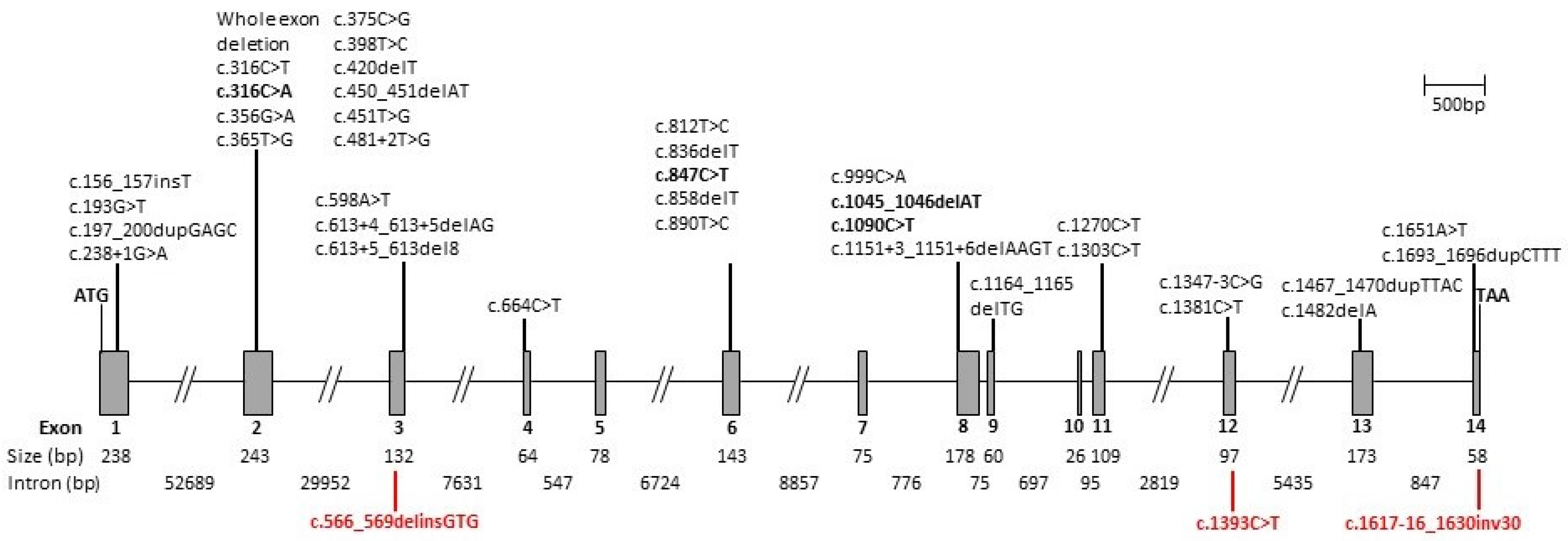
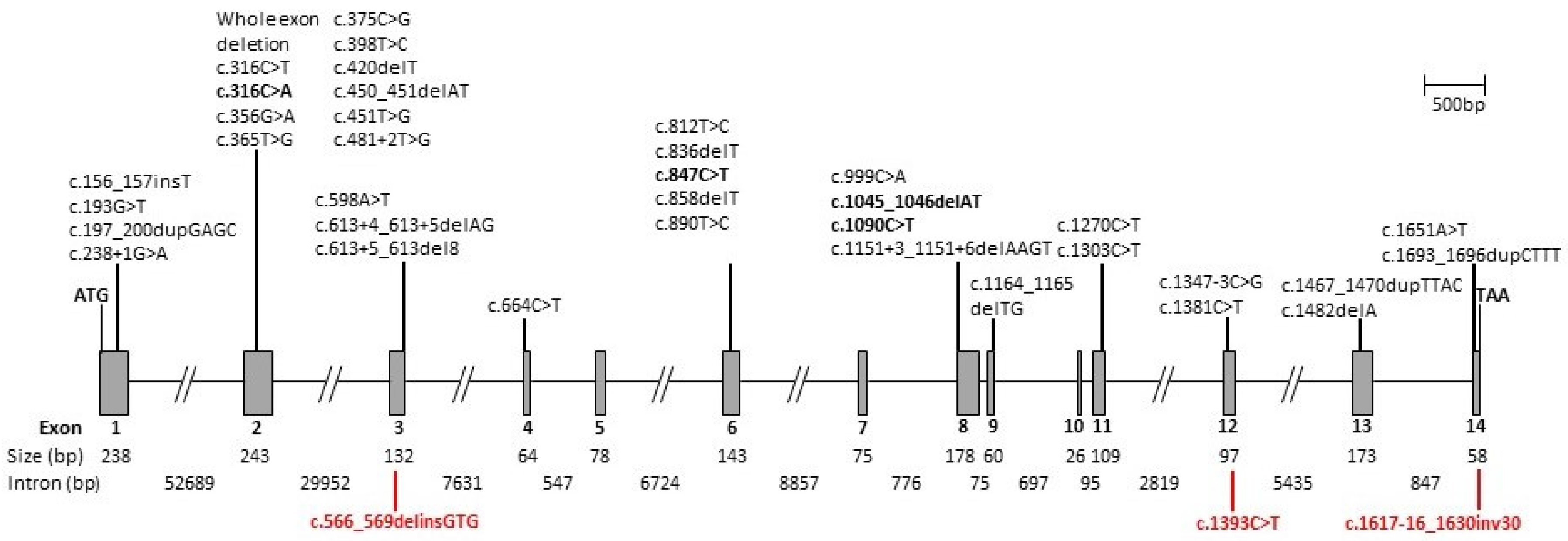
| Variant | Effect | References | ||
|---|---|---|---|---|
| c.156_157insT | Exon 1 | GAG > TGA | p.(Glu53*) | [15] |
| c.193G > T | Exon 1 | GAG > TAG | p.(Glu65*) | [10,16] |
| c.197_200dupGAGC | Exon 1 | CTG > AGC | p.(Leu68Serfs*15) | [17,18] |
| c.238 + 1G > A | Intron 1 | Splicing defect | [11,19] | |
| Whole exon deletion | Exons 1 and 2 | Loss of function | [20] | |
| Whole exon deletion | Exon 2 | Loss of function | [17] | |
| c.316C > T | Exon 2 | CGT > TGT | p.(Arg106Cys) | [10,16] |
| c.316C > A | Exon 2 | CGT > AGT | p.(Arg106Ser) | [18,21] This report |
| c.356G > A | Exon 2 | GGT > GAT | p.(Gly119Asp) | [22] |
| c.365T > G | Exon 2 | CTC > CGC | p.(Leu122Arg) | [23] |
| c.375C > G | Exon 2 | TGC > TGG | p.(Cys125Trp) | [10,16,24] |
| c.398T > C | Exon 2 | CTA > CCA | p.(Leu133Pro) | [25] |
| c.420delT | Exon 2 | CTT > CTA | p.(Leu140Leufs*2) | [26] |
| c.450_451delAT | Exon 2 | ATA > ATG | p.(Ile150Metfs*3) | [27] |
| c.451T > G | Exon 2 | TGG > GGG | p.(Trp151Gly) | [28] |
| c.481 + 2T > G | Intron 2 | Splicing defect | [29] | |
| c.566_569delinsGTG | Exon 3 | p.(Lys189Serfs*5) | This report | |
| c.598A > T | Exon 3 | AAA > TAA | p.(Lys200*) | [26,30] |
| c.613 + 4_613 + 5delAG | Intron 3 | Splicing defect | [18] | |
| c.613 + 5_613del8 | Intron 3 | Splicing defect | [31] | |
| c.664C > T | Exon 4 | CAG > TAG | p.(Gln222*) | [32] |
| c.812T > C | Exon 6 | CTG > CCG | p.(Leu271Pro) | [30,33] |
| c.836delT | Exon 6 | ATG > AGG | p.(Met279Argfs*6) | [15,28] |
| c.847C > T | Exon 6 | CGA > TGA | p.(Arg283*) | [4,10,13,16,17,18,22,24,26,30,32,34,35,36,37,38,39,40,41,42,43] This report |
| c.858delT | Exon 6 | ACT > ACC | p.(Thr260Thrfs*10) | [26] |
| c.890T > C | Exon 6 | ATA > ACA | p.(Ile297Thr) | [19,30] |
| c.899-1G > A | Intron 6 | Splicing defect | [43] | |
| c.999C > A | Exon 8 | TGC > TGA | p.(Cys333*) | [44] |
| c.1045_1046delAT | Exon 8 | ATG > GTT | p.(Met349Valfs*20) | [16,43,45] This report |
| c.1090C > T | Exon 8 | CGA > TGA | p.(Arg364*) | [22,46] This report |
| c.1151 + 3_1151 + 6delAAGT | Intron 8 | Splicing defect | [12,18] | |
| c.1164_1165delTG | Exon 9 | TGT > TGA | p.(Cys388*) | [27,36] |
| c.1270C > T | Exon 11 | CAG > TAG | p.(Gln424*) | [29] |
| c.1303C > T | Exon11 | CGA > TGA | p.(Arg435*) | [39] |
| c.1347 − 3C > G | Intron 11 | Splicing defect | [18] | |
| c.1381C > T | Exon 12 | CGA > TGA | p.(Arg461*) | [38] |
| c.1393C > T | Exon 12 | CGG > TGG | p.(Arg465Trp) | This report |
| c.1467_1470dupTTAC | Exon 13 | ACT > TTA | p.(Thr491Leufs*4) | [47] |
| c.1482delA | Exon 13 | GAA > GAG | p.(Glu494Glufs*1) | [28,48] |
| c.1617-16_1630inv30 | Partial intron 13/exon 14 | This report | ||
| c.1651A > T | Exon 14 | AGC > TGC | p.(Ser551Cys) | [17] |
| c.1693_1696dupCTTT | Exon 14 | TAT > TCT | p.(Tyr548Serfs*19) | [25] |
| Patient | Gender | Ethnicity | Symptoms at Presentation | Age at First Presentation | Age at Last Visit | Lens Status | Visual Acuity (logMAR) at Last Visit | Visual Fields (Horizontal Loss) | Fundoscopy | Variant | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | OD | OS | OD | OS | ||||||||
| A | M | White Caucasian (Irish) | ↓ colour vision nyctalopia | 25 | 35 | N | N | 6/24 | HM | NP | NP | RP phenotype with widespread pigment, disc pallor, attenuated vessels and central atrophy sparing the fovea. | p.(Arg283*) Hom |
| B | F | Indian | ↓ central vision slow dark adaptation | 45 | 60 | N | N | 2/60 | 6/36 | Central loss extending to 10–30° | Central loss extending to 20–30° | No pigment, central atrophy with foveal sparing. | p.(Arg283*) p.(Arg364*) |
| C | M | White Caucasian | ↓ central vision, photopsia, nyctalopia and field loss | 20 | 56 | Pseudophakic | Pseudophakic | NPL | NPL | NP | NP | RP phenotype with widespread pigment, disc pallor, attenuated vessels and central atrophy with strip of foveal sparing. | p.(Arg283*) p.(Lys189Serfs*5) |
| D | F | Pakistani | Photosensitivity ↓ colour vision ↓ central vision field loss, photopsia, nyctalopia | 25 | 30 | N | N | 6/18 | 6/12 | <10° | <10° | Sparse pigmentation, disc pallor, attenuated vessels; distinct nummular RPE depigmentation in the mid-periphery with chorioretinal atrophic patches temporally. | c.1617-16_1630inv30 Hom |
| E | M | Indian | ↓ central vision, nyctalopia | 7 | 14 | PSCLO | PSCLO | POL | POL | NP | NP | Dense pigmentation throughout the fundus, disc pallor, significantly attenuated vessels; widespread atrophy throughout fundus. | p.(Met349Valfs*20) Hom |
| F | F | Kashmiri | ↓ central vision | 40 | 57 | N | N | 6/60 | 6/60 | NP | NP | No pigment, central atrophy, normal colour discs. | p.(Arg465Trp) p.(Arg106Ser) |
| Patient | Autofluorescence | CMT OD (μm) | CMT OS (μm) | OCT |
|---|---|---|---|---|
| A | Well demarcated dense loss of signal centrally, less dense atrophy external to arcades; increased focal AF signal at foveae, surrounded by atrophy within maculae. | 266 | 237 | Significant loss of ellipsoid zone in central retina with foveal sparing at site of increased AF signal; thin retinas, epiretinal changes. |
| B | Atrophy primarily confined to macular region with concentric patchy loss of signal and increased signal extending to arcades; foveal preservation. | 229 | 266 | Significant loss of outer retinal layers in macular region, with central foveal preservation in remnant corresponding to AF increased signal. |
| C | Loss of signal consistent with central atrophy with a strip of minimal central sparing. There were also peripheral areas of loss-of-signal consistent with the chorioretinal atrophic patches. | 210 | 193 | Loss of outer retina bilaterally. |
| D | Increased signal in the maculae surrounding the foveal atrophy, with patchy, punctate loss extending from and including the arcades. | 176 | 183 | Focal foveal disruption of ellipsoid layer and loss of the outer retinal layers in both eyes. |
| E | Complete loss of signal consistent with coalescing patches of atrophy throughout the fundi, including both maculae. | 279 | 285 | Loss of outer retina bilaterally with thickened inner retina. |
| F | AF imaging showed significant central loss of signal denoting atrophy with a thick band of increased AF extending to the disc and around the circumference of the atrophy. | 126 | 131 | Significant outer retinal loss in both eyes. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Downes, S.M.; Nguyen, T.; Tai, V.; Broadgate, S.; Shah, M.; Al-Khuzaei, S.; MacLaren, R.E.; Shanks, M.; Clouston, P.; Halford, S. Genetic and Clinical Findings in an Ethnically Diverse Cohort with Retinitis Pigmentosa Associated with Pathogenic Variants in CERKL. Genes 2020, 11, 1497. https://doi.org/10.3390/genes11121497
Downes SM, Nguyen T, Tai V, Broadgate S, Shah M, Al-Khuzaei S, MacLaren RE, Shanks M, Clouston P, Halford S. Genetic and Clinical Findings in an Ethnically Diverse Cohort with Retinitis Pigmentosa Associated with Pathogenic Variants in CERKL. Genes. 2020; 11(12):1497. https://doi.org/10.3390/genes11121497
Chicago/Turabian StyleDownes, Susan M., Tham Nguyen, Vicky Tai, Suzanne Broadgate, Mital Shah, Saoud Al-Khuzaei, Robert E. MacLaren, Morag Shanks, Penny Clouston, and Stephanie Halford. 2020. "Genetic and Clinical Findings in an Ethnically Diverse Cohort with Retinitis Pigmentosa Associated with Pathogenic Variants in CERKL" Genes 11, no. 12: 1497. https://doi.org/10.3390/genes11121497
APA StyleDownes, S. M., Nguyen, T., Tai, V., Broadgate, S., Shah, M., Al-Khuzaei, S., MacLaren, R. E., Shanks, M., Clouston, P., & Halford, S. (2020). Genetic and Clinical Findings in an Ethnically Diverse Cohort with Retinitis Pigmentosa Associated with Pathogenic Variants in CERKL. Genes, 11(12), 1497. https://doi.org/10.3390/genes11121497