The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein

 ,
, 
 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Clinical Exome Sequencing (CES)
2.3. SNP-Array Analysis
3. Results
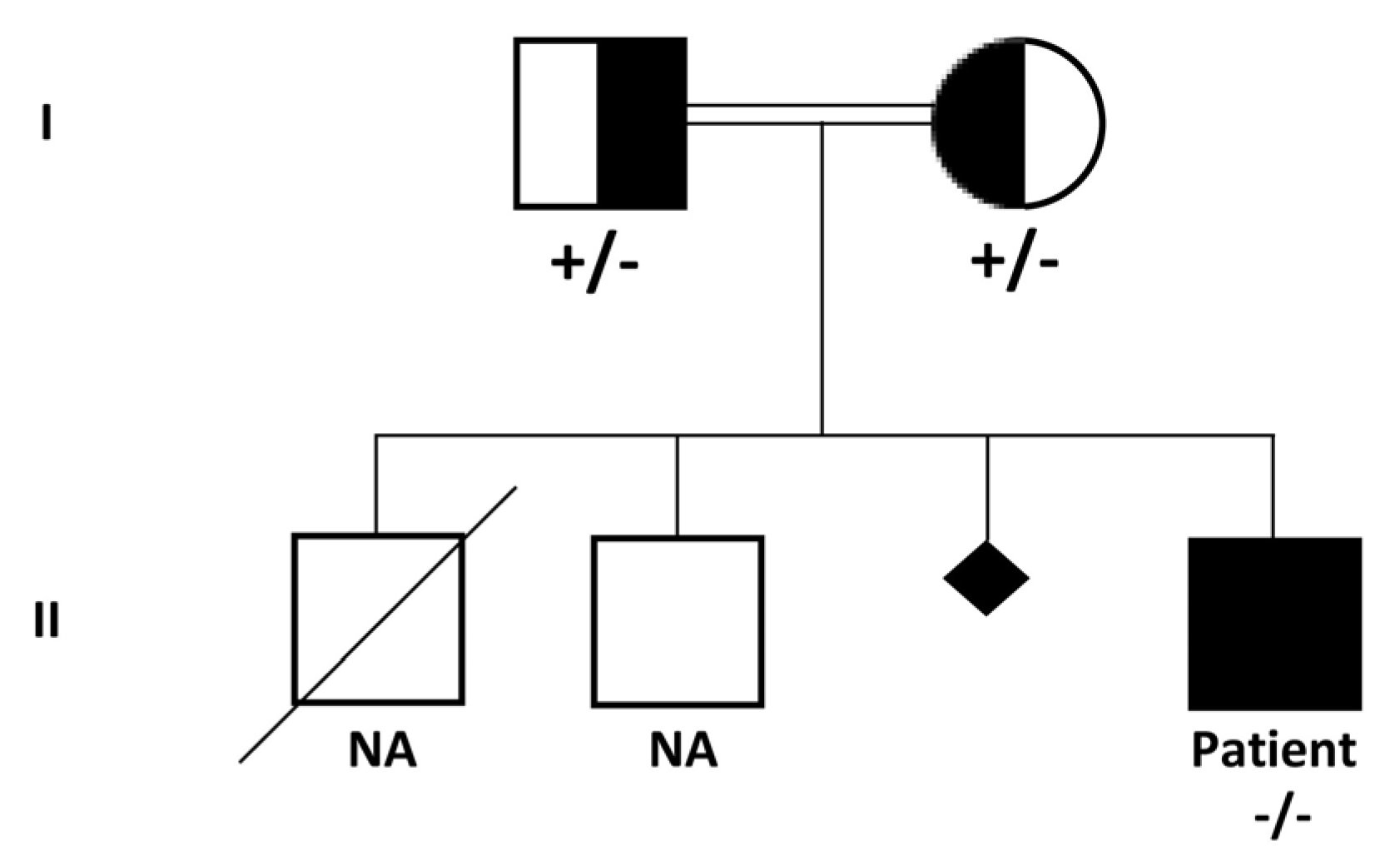
3.1. Clinical History
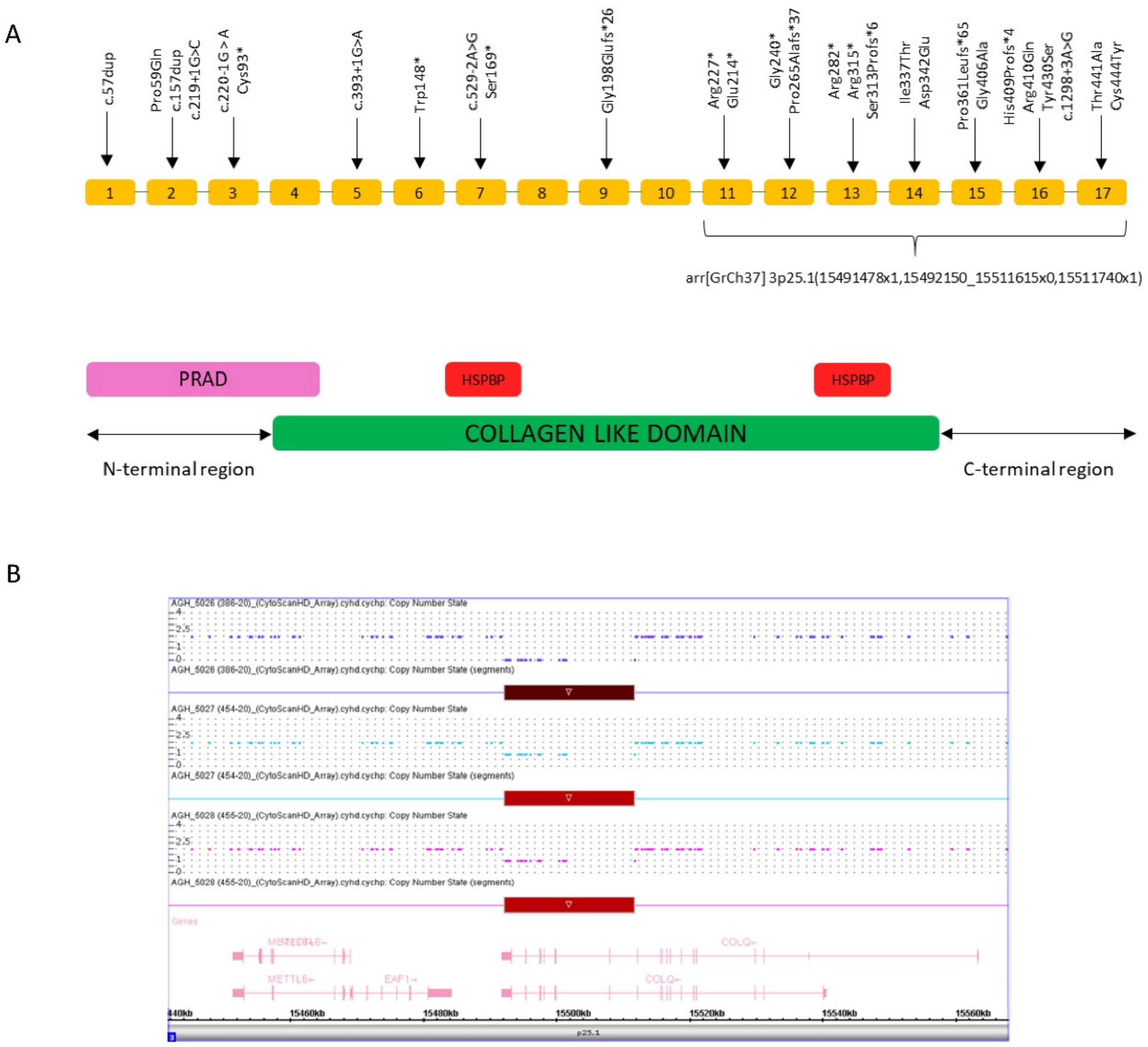
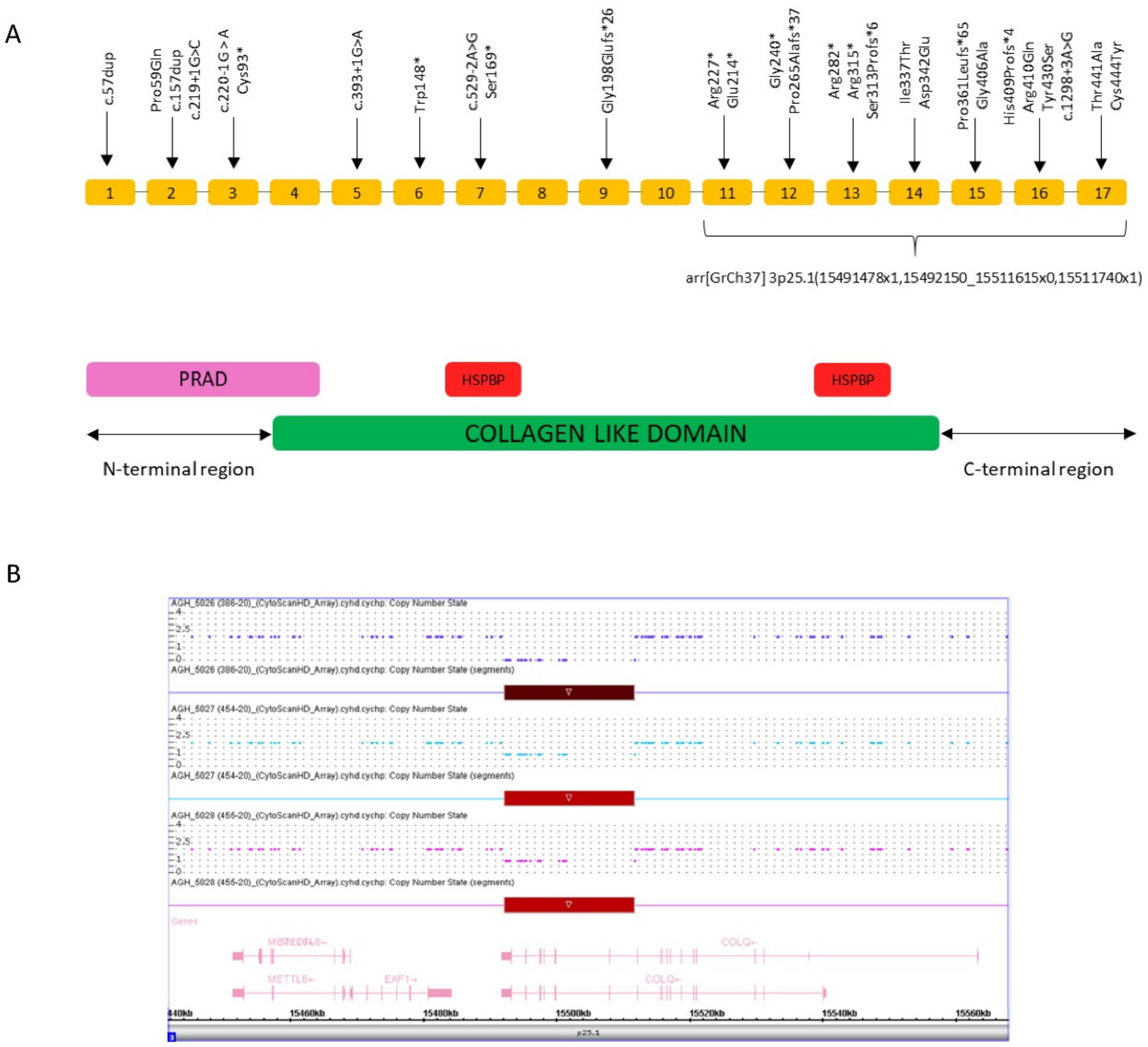
3.2. Genetic Findings
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Bernasconi, P.; D’Amico, A.; Brugnoni, R.; Fiorillo, C.; Garibaldi, M.; Astrea, G.; Bruno, C.; Santorelli, F.M.; Liguori, R.; et al. Italian recommendations for diagnosis and management of congenital myasthenic syndromes. Neurol. Sci. 2019, 40, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.-M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- De Souza, P.V.S.; Batistella, G.N.D.R.; Lino, V.C.; Pinto, W.B.V.D.R.; Annes, M.; Oliveira, A.S.B. Clinical and genetic basis of congenital myasthenic syndromes. Arq. Neuro-Psiquiatria 2016, 74, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.M.R.; Palace, J.; Beeson, D. Inherited disorders of the neuromuscular junction: An update. J. Neurol. 2014, 261, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Ardissone, A.; Moroni, I.; Bernasconi, P.; Brugnoni, R. Congenital myasthenic syndrome: Phenotypic variability in patients harbouring p.T159P mutation in CHRNE gene. Acta Myol. 2017, 36, 28–32. [Google Scholar] [PubMed]
- Palumbo, O.; Fichera, M.; Palumbo, P.; Rizzo, R.; Mazzolla, E.; Cocuzza, D.M.; Carella, M.; Mattina, T. TBR1 is the candidate gene for intellectual disability in patients with a 2q24.2 interstitial deletion. Am. J. Med Genet. Part A 2014, 164, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, O.; Palumbo, P.; Di Muro, E.; Cinque, L.; Petracca, A.; Carella, M.; Castori, M. A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features. Genes 2020, 11, 707. [Google Scholar] [CrossRef] [PubMed]
- Barisic, N.; Müller, J.; Paucic-Kirincic, E.; Gazdik, M.; Lah-Tomulic, K.; Pertl, A.; Sertić, J.; Zurak, N.; Lochmüller, H.; Abicht, A. Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. Eur. J. Paediatr. Neurol. 2005, 9, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Byring, R.; Pihko, H.; Tsujino, A.; Shen, X.-M.; Gustafsson, B.; Hackman, P.; Ohno, K.; Engel, A.G.; Udd, B. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul. Disord. 2002, 12, 548–553. [Google Scholar] [CrossRef]
- Abicht, A.; Müller, J.S.; Lochmüller, H. Congenital Myasthenic Syndromes; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mefford, H.C., Stephens, K., Amemiya, A., Ledbetter, N., Eds.; GeneReviews; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Arredondo, J.; Lara, M.; Ng, F.; Gochez, D.A.; Lee, D.C.; Logia, S.P.; Nguyen, J.; Maselli, R.A. COOH-terminal collagen Q (COLQ) mutants causing human deficiency of endplate acetylcholinesterase impair the interaction of ColQ with proteins of the basal lamina. Hum. Genet. 2014, 133, 599–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, D.O.; Walls, T.J.; Nakano, S.; Camp, S.; Taylor, P.; Harper, C.M.; Groover, R.V.; Peterson, H.A.; Jamieson, D.G.; Engel, A.G. Congenital endplate acetylcholinesterase deficiency. Brain 1993, 116, 633–653. [Google Scholar] [CrossRef] [PubMed]
- Al-Shahoumi, R.; Brady, L.I.; Schwartzentruber, J.; Tarnopolsky, M. Two cases of congenital myasthenic syndrome with vocal cord paralysis. Neurology 2015, 84, 1281–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, K.; Brengman, J.; Tsujino, A.; Engel, A.G. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 9654–9659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wu, Y.; Wang, C.; Jiao, J.; Klein, C.J. Copy number analysis reveals a novel multiexon deletion of the COLQ gene in congenital myasthenia. Neurol. Genet. 2016, 2, e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreux, F.; Hantaï, D.; Eymard, B. Congenital myasthenic syndromes phenotypic expression and pathophysiological characterisation. Rev. Neurol. 2004, 160, 163–176. [Google Scholar] [CrossRef]
- Padmanabha, H.; Saini, A.G.; Sankhyan, N.; Singhi, P. COLQ-Related Congenital Myasthenic Syndrome and Response to Salbutamol Therapy. J. Clin. Neuromuscul. Dis. 2017, 18, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, A.; Müller, J.S.; Guergueltcheva, V.; Dusl, M.; Schara, U.; Rakočević-Stojanović, V.; Lindberg, C.; Scola, R.H.; Werneck, L.C.; Colomer, J.; et al. A retrospective clinical study of the treatment of slow-channel congenital myasthenic syndrome. J. Neurol. 2012, 259, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Ng, J.J.; A Anderson, J.; Cagney, O.; Arredondo, J.; Williams, C.; Wessel, H.B.; Abdel-Hamid, H.; Wollmann, R.L. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J. Med Genet. 2009, 46, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laforgia, N.; De Cosmo, L.; Palumbo, O.; Ranieri, C.; Sesta, M.; Capodiferro, D.; Pantaleo, A.; Iapicca, P.; Lastella, P.; Capozza, M.; et al. The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein. Genes 2020, 11, 1519. https://doi.org/10.3390/genes11121519
Laforgia N, De Cosmo L, Palumbo O, Ranieri C, Sesta M, Capodiferro D, Pantaleo A, Iapicca P, Lastella P, Capozza M, et al. The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein. Genes. 2020; 11(12):1519. https://doi.org/10.3390/genes11121519
Chicago/Turabian StyleLaforgia, Nicola, Lucrezia De Cosmo, Orazio Palumbo, Carlotta Ranieri, Michela Sesta, Donatella Capodiferro, Antonino Pantaleo, Pierluigi Iapicca, Patrizia Lastella, Manuela Capozza, and et al. 2020. "The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein" Genes 11, no. 12: 1519. https://doi.org/10.3390/genes11121519
APA StyleLaforgia, N., De Cosmo, L., Palumbo, O., Ranieri, C., Sesta, M., Capodiferro, D., Pantaleo, A., Iapicca, P., Lastella, P., Capozza, M., Schettini, F., Bukvic, N., Bagnulo, R., & Resta, N. (2020). The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein. Genes, 11(12), 1519. https://doi.org/10.3390/genes11121519