Transcriptomic and Metabolomic Changes Triggered by Fusarium solani in Common Bean (Phaseolus vulgaris L.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Growth and in Vivo Inoculations
2.2. RNA Extraction, Construction of Illumina Library, and Sequencing
2.3. Sequencing Data Analysis
2.4. Differential Expression, Enrichment Analysis, and Real-Time qRT-PCR
2.5. Metabolome Analysis
2.5.1. Sample Preparation
2.5.2. Chromatographic Mass Spectrometry Acquisition Conditions
2.5.3. Metabolomics Data Analysis
2.6. Co-Joint Analysis of Transcriptome and Metabolome
3. Results
3.1. Phenotypic Performance of Common Bean Roots under FSP Infection
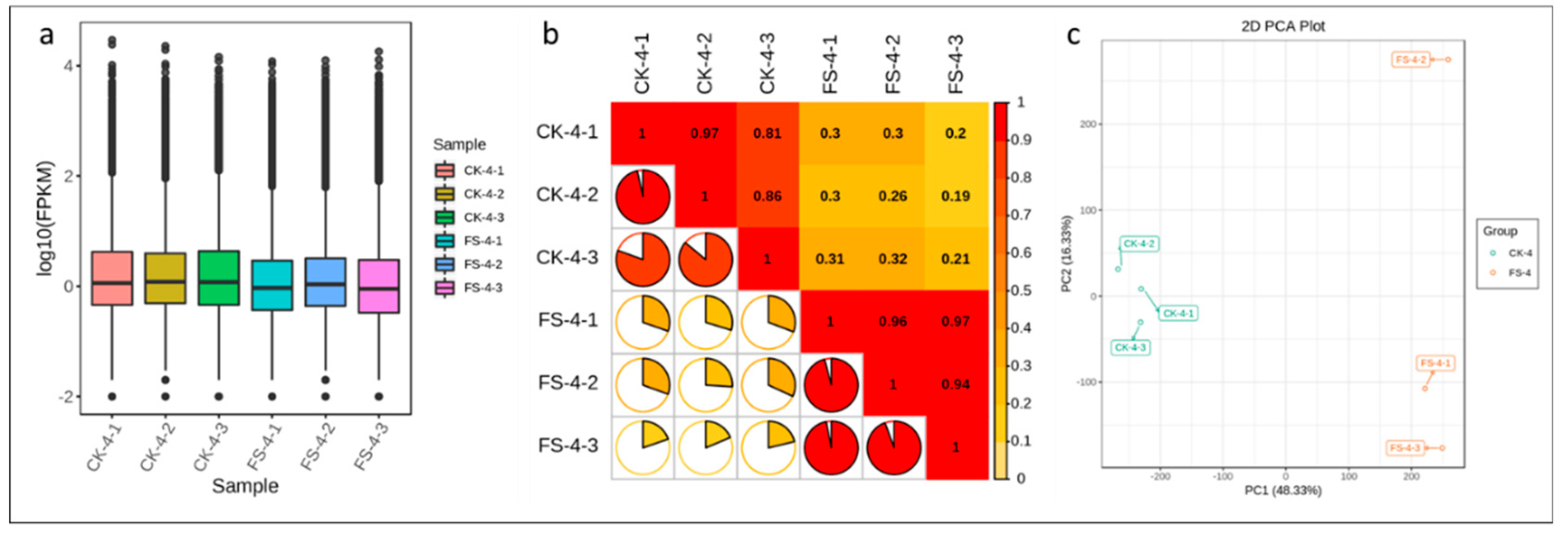
3.2. Overview of Transcriptomic Analysis
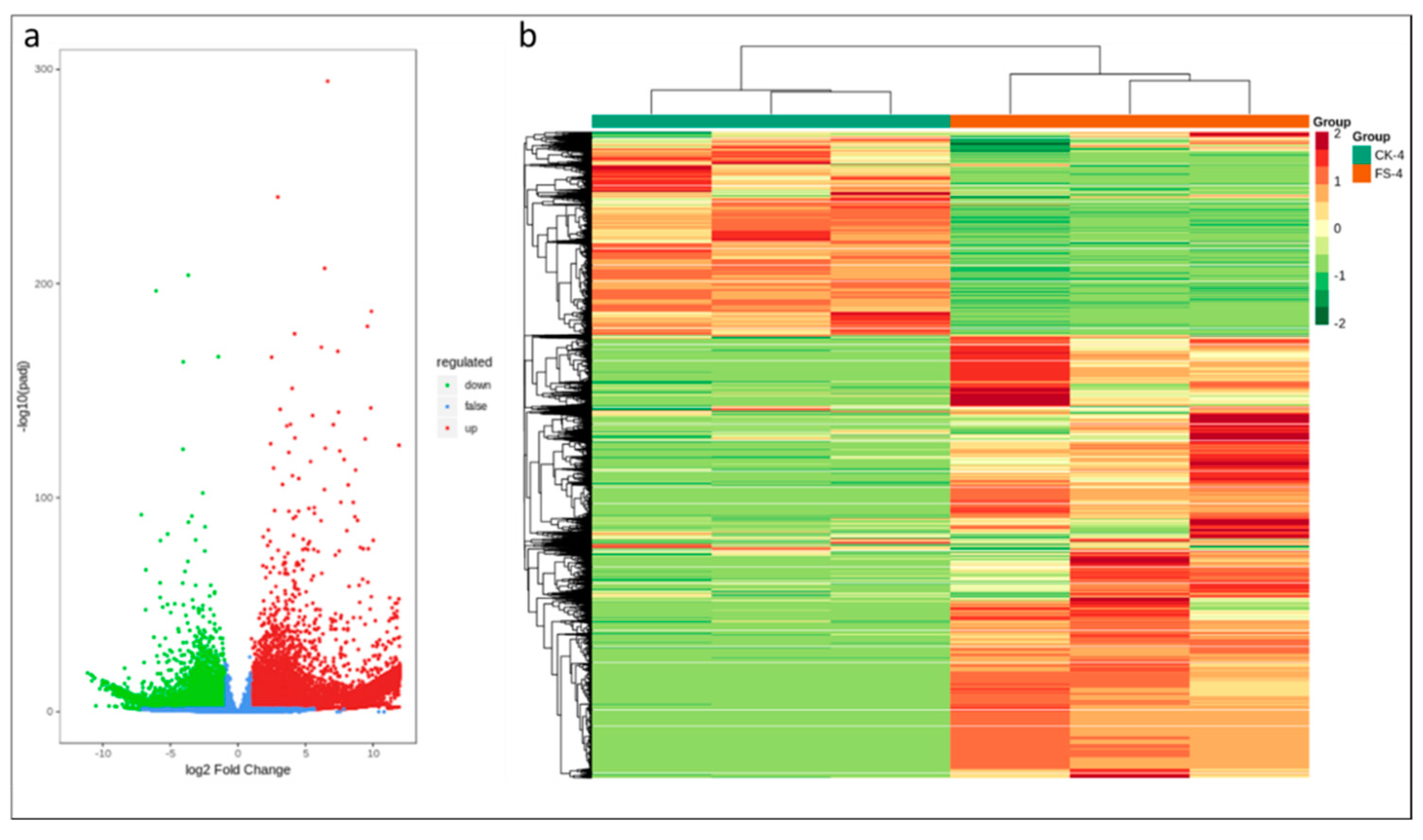
3.3. Differential Gene Expression Analysis
3.4. Transcriptomic Response of FSP-Infected Common Bean Roots
3.4.1. FSP Perception
3.4.2. Signal Transduction
Activation of Signaling Mechanisms
Systematic Acquired Resistance by Hormones
3.4.3. ROS and Cell Wall Modification of Common Bean Roots After FSP infection
3.5. Validation of DEGs by qRT-PCR
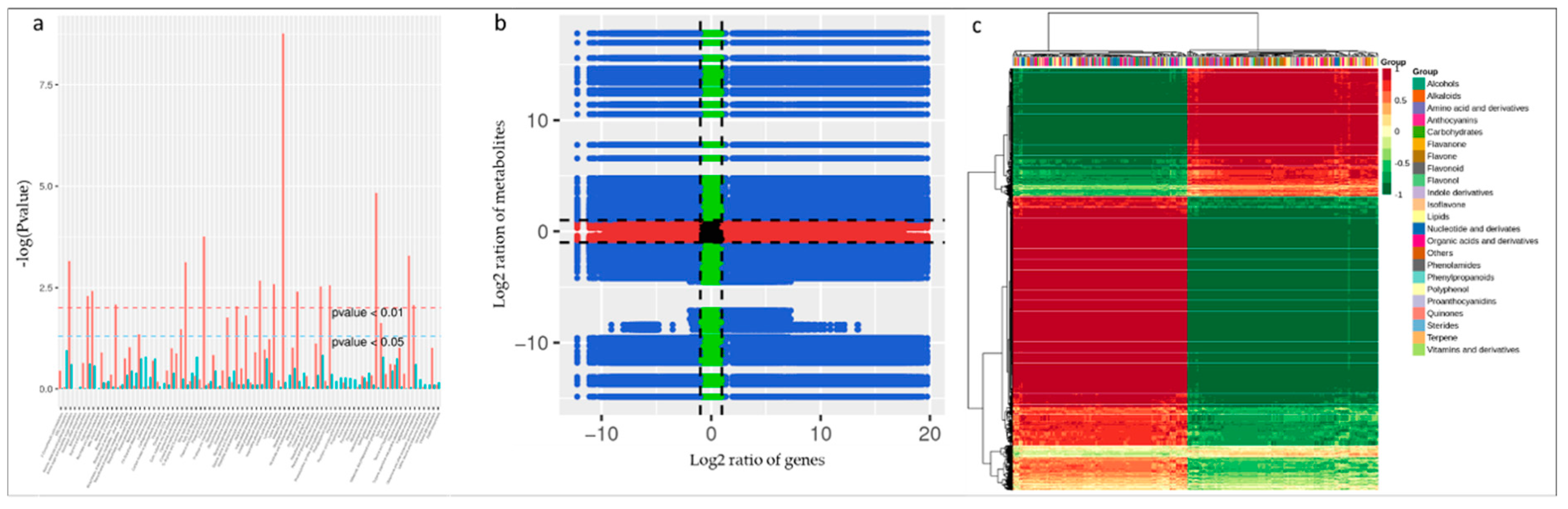
3.6. Metabolomic Response of FS-Infected Common Bean Roots
4. Discussion
4.1. FSP is Perceived by PRRs, WAKs, and NLRs, Which Activate Multiple Signaling Mechanisms
4.2. FSP Infection Triggered ROS Production in Common Bean Roots
4.3. Cell Wall Acts as a Barrier Against FSP Infection in Common Bean
4.4. FSP Infection Triggered Different Metabolic Responses in Common Bean
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, L.; Yang, T.; Liu, R.; Redden, B.; Maalouf, F.; Zong, X. Food legume production in China. Crop J. 2017, 5, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Macedo, R.; Sales, L.P.; Yoshida, F.; Silva-Abud, L.L.; Junior, M.L. Potential worldwide distribution of Fusarium dry root rot in common beans based on the optimal environment for disease occurrence. PLoS ONE 2017, 12, e0187770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongom, P.; Rubaihayo, P.; Nkalubo, S.; Gibson, P. Inheritance of resistance to Fusarium root rot in three common bean genotypes. Electron. J. Plant Breed. 2012, 3, 892–897. [Google Scholar]
- Wang, W.; Jacobs, J.L.; Chilvers, M.I.; Mukankusi, C.M.; Kelly, J.D.; Cichy, K.A. QTL analysis of Fusarium root rot resistance in an Andean× middle American common bean RIL population. Crop Sci. 2018, 58, 1166–1180. [Google Scholar] [CrossRef]
- Mukankusi, C.; Derera, J.; Melis, R.; Gibson, P.T.; Buruchara, R. Genetic analysis of resistance to Fusarium root rot in common bean. Euphytica 2011, 182, 11. [Google Scholar] [CrossRef]
- Andersen, E.J.; Ali, S.; Byamukama, E.; Yen, Y.; Nepal, M.P. Disease resistance mechanisms in plants. Genes 2018, 9, 339. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Liu, N.; Li, C.; Wang, X.; Xu, X.; Chen, W.; Xing, G.; Zheng, W. The early response during the interaction of fungal phytopathogen and host plant. Open Biol. 2017, 7, 170057. [Google Scholar] [CrossRef] [Green Version]
- Román-Avilés, B.; Kelly, J.D. Identification of quantitative trait loci conditioning resistance to Fusarium root rot in common bean. Crop Sci. 2005, 45, 1881–1890. [Google Scholar] [CrossRef] [Green Version]
- D’Ippólito, S.; Martín, M.L.; Salcedo, M.F.; Atencio, H.M.; Casalongué, C.A.; Godoy, A.V.; Fiol, D.F. Transcriptome profiling of Fusarium solani f. sp. eumartii-infected potato tubers provides evidence of an inducible defense response. Physiol. Mol. Plant Pathol. 2010, 75, 3–12. [Google Scholar] [CrossRef]
- Hadwiger, L.A. Anatomy of a nonhost disease resistance response of pea to Fusarium solani: PR gene elicitation via DNase, chitosan and chromatin alterations. Front. Plant Sci. 2015, 6, 373. [Google Scholar] [CrossRef] [PubMed]
- Bawa, G.; Feng, L.; Li, Y.; Shang, J.; Wu, X.; Chang, X.; Sun, X.; Yu, L.; Liu, C.; Du, J.; et al. Physiological Analysis Reveals the Possible Resistance Mechanisms of Glycine max to Fusarium solani. J. Agric. Sci. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhao, Q.; Cui, X.; Chen, R.; Li, X.; Qiu, B.; Ge, F. A transcriptome analysis uncovers Panax notoginseng resistance to Fusarium solani induced by methyl jasmonate. Genes Genom. 2019, 41, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kim, J.-H.; Jung, B.; Ji, S.; Seo, M.W.; Han, Y.K.; Lee, S.W.; Bae, Y.S.; Choi, H.-G.; Lee, S.-H.; et al. Transcriptome analyses of the ginseng root rot pathogens Cylindrocarpon destructans and Fusarium solani to identify radicicol resistance mechanisms. J. Ginseng Res. 2018. [Google Scholar] [CrossRef]
- Chen, F.; Ma, R.; Chen, X.-L. Advances of Metabolomics in Fungal Pathogen–Plant Interactions. Metabolites 2019, 9, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wu, Q.; He, W.; He, T.; Wu, Q.; Miao, Y. Combined De Novo Transcriptome and Metabolome Analysis of Common Bean Response to Fusarium oxysporum f. sp. phaseoli Infection. Int. J. Mol. Sci. 2019, 20, 6278. [Google Scholar] [CrossRef] [Green Version]
- Coppola, M.; Diretto, G.; Digilio, M.C.; Woo, S.L.; Giuliano, G.; Molisso, D.; Lorito, M.; Pennacchio, F.; Rao, R. Transcriptome and metabolome reprogramming in tomato plants by Trichoderma harzianum strain T22 primes and enhances defence responses against aphids. Front. Physiol. 2019, 10, 745. [Google Scholar] [CrossRef]
- Ye, W.; Liu, T.; Zhang, W.; Li, S.; Zhu, M.; Li, H.; Kong, Y.; Xu, L. Disclosure of the Molecular Mechanism of Wheat Leaf Spot Disease Caused by Bipolaris sorokiniana through Comparative Transcriptome and Metabolomics Analysis. Int. J. Mol. Sci. 2019, 20, 6090. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Z.; Fang, W.; Ma, X.; Chen, T.; Cui, G.; Ma, Y.; Kang, L.; Nan, T.; Lin, H.; Tang, J.; et al. Metabolome and transcriptome analyses reveal quality change in the orange-rooted Salvia miltiorrhiza (Danshen) from cultivated field. Chin. Med. 2019, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.; Lu, X.; Chen, A.; Luo, Y.; Hao, J.; Gao, W. Effects of Fusarium solani and F. oxysporum Infection on the Metabolism of Ginsenosides in American Ginseng Roots. Molecules 2015, 20, 10535–10552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.I.; Muthusamy, M.; Nawaz, M.A.; Hong, J.K.; Lim, M.-H.; Kim, J.A.; Jeong, M.-J. Genome-wide analysis of spatiotemporal gene expression patterns during floral organ development in Brassica rapa. Mol. Genet. Genom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Jeon, J.; Park, S.; Kim, K.; Choi, G.; Lee, H.; Kim, Y.; Yang, H.; Yeo, J.; Lee, Y.; et al. Genome seqeunce of an endophytic fungus, Fusarium solani JS-169, which has antifungal activity. Genome Announc. 2017, 5, e01071-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG Database: A tool for fenome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Profile hidden markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protocol. 2013, 8, 1765. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Xue, R.; Wu, J.; Zhu, Z.; Wang, L.; Wang, X.; Wang, S.; Blair, M.W. Differentially expressed genes in resistant and susceptible common bean (Phaseolus vulgaris L.) genotypes in response to Fusarium oxysporum f. sp. phaseoli. PLoS ONE 2015, 10, e0127698. [Google Scholar] [CrossRef] [PubMed]
- Silvia Sebastiani, M.; Bagnaresi, P.; Sestili, S.; Biselli, C.; Zechini, A.; Orrù, L.; Cattivelli, L.; Ficcadenti, N. Transcriptome analysis of the melon-Fusarium oxysporum f. sp. melonis race 1.2 pathosystem in susceptible and resistant plants. Front. Plant Sci. 2017, 8, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Ying, H.; Pingcuo, G.; Wang, S.; Zhao, F.; Cui, Y.; Shi, J.; Zeng, H.; Zeng, X. Identification of Potential Metabolites Mediating Bird’s Selective Feeding on Prunus mira Flowers. BioMed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography—Mass spectrometry, XCMS, and chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]
- Urbanczyk-Wochniak, E.; Luedemann, A.; Kopka, J.; Selbig, J.; Roessner-Tunali, U.; Willmitzer, L.; Fernie, A.R. Parallel analysis of transcript and metabolic profiles: A new approach in systems biology. EMBO Rep. 2003, 4, 989–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miya, A.; Albert, P.; Shinya, T.; Desaki, Y.; Ichimura, K.; Shirasu, K.; Narusaka, Y.; Kawakami, N.; Kaku, H.; Shibuya, N. CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 19613–19618. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Zhang, X.-C.; Neece, D.; Ramonell, K.M.; Clough, S.; Kim, S.-Y.; Stacey, M.G.; Stacey, G. A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 2008, 20, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Brar, A.; Yadav, M.; Chawade, A.; Vivekanand, V.; Pareek, N. Chitinases—Potential candidates for enhanced plant resistance towards fungal pathogens. Agriculture 2018, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, C. Plant pattern-recognition receptors. Trends Immunol. 2014, 35, 345–351. [Google Scholar] [CrossRef]
- Ali, M.A.; Anjam, M.S.; Nawaz, M.A.; Lam, H.-M.; Chung, G. Signal transduction in plant–nematode interactions. Int. J. Mol. Sci. 2018, 19, 1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Shah, J.; Klessig, D.F. Signal perception and transduction in plant defense responses. Genes Dev. 1997, 11, 1621–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, M.A.; Rehman, H.M.; Imtiaz, M.; Baloch, F.S.; Lee, J.D.; Yang, S.H.; Lee, S.I.; Chung, G. Systems Identification and Characterization of Cell Wall Reassembly and Degradation Related Genes in Glycine max (L.) Merill, a Bioenergy Legume. Sci. Rep. 2017, 7, 10862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caño-Delgado, A.; Penfield, S.; Smith, C.; Catley, M.; Bevan, M. Reduced cellulose synthesis invokes lignification and defense responses in Arabidopsis thaliana. Plant J. 2003, 34, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, V.; Vashisht, D.; Cletus, J.; Sakthivel, N. Plant β-1, 3-glucanases: Their biological functions and transgenic expression against phytopathogenic fungi. Biotechnol. Lett. 2012, 34, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [Green Version]
- Kärkönen, A.; Kuchitsu, K. Reactive oxygen species in cell wall metabolism and development in plants. Phytochemistry 2015, 112, 22–32. [Google Scholar] [CrossRef]
- Cao, H.; Ji, Y.; Li, S.; Lu, L.; Tian, M.; Yang, W.; Li, H. Extensive metabolic profiles of leaves and stems from the medicinal plant Dendrobium officinale Kimura et Migo. Metabolites 2019, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Kaku, H.; Nishizawa, Y.; Ishii-Minami, N.; Akimoto-Tomiyama, C.; Dohmae, N.; Takio, K.; Minami, E.; Shibuya, N. Plant cells recognize chitin fragments for defense signaling through a plasma membrane receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 11086–11091. [Google Scholar] [CrossRef] [Green Version]
- Decreux, A.; Messiaen, J. Wall-associated kinase WAK1 interacts with cell wall pectins in a calcium-induced conformation. Plant Cell Physiol. 2005, 46, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, N.; Inoue, H.; Kato, T.; Funao, T.; Shirota, M.; Shimizu, T.; Kanamori, H.; Yamane, H.; Hayano-Saito, Y.; Matsumoto, T.; et al. Durable panicle blast-resistance gene Pb1 encodes an atypical CC-NBS-LRR protein and was generated by acquiring a promoter through local genome duplication. Plant J. 2010, 64, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Mentlak, T.A.; Kombrink, A.; Shinya, T.; Ryder, L.S.; Otomo, I.; Saitoh, H.; Terauchi, R.; Nishizawa, Y.; Shibuya, N.; Thomma, B.P.; et al. Effector-mediated suppression of chitin-triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. Plant Cell 2012, 24, 322–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robatzek, S. Endocytosis: At the crossroads of pattern recognition immune receptors and pathogen effectors. In Applied Plant Cell Biology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 273–297. [Google Scholar]
- Frye, C.A.; Tang, D.; Innes, R.W. Negative regulation of defense responses in plants by a conserved MAPKK kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 373–378. [Google Scholar] [CrossRef]
- Felix, G.; Grosskopf, D.G.; Regenass, M.; Boller, T. Rapid changes of protein phosphorylation are involved in transduction of the elicitor signal in plant cells. Proc. Natl. Acad. Sci. USA 1991, 88, 8831–8834. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, F.; Mizoguchi, T.; Yoshida, R.; Ichimura, K.; Shinozaki, K. Calmodulin-dependent activation of MAP kinase for ROS homeostasis in Arabidopsis. Mol. Cell 2011, 41, 649–660. [Google Scholar] [CrossRef]
- Temple, B.R.; Jones, A.M. The plant heterotrimeric G-protein complex. Annu. Rev. Plant Biol. 2007, 58, 249–266. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gao, Z.; Zheng, X.; Zhang, Z. The role of G-proteins in plant immunity. Plant Signal. Behav. 2012, 7, 1284–1288. [Google Scholar] [CrossRef]
- Gupta, S.; Chakraborti, D.; Sengupta, A.; Basu, D.; Das, S. Primary metabolism of chickpea is the initial target of wound inducing early sensed Fusarium oxysporum f. sp. ciceri race I. PLoS ONE 2010, 5, e9030. [Google Scholar] [CrossRef]
- Bari, R.; Jones, J.D.G. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Passardi, F.; Cosio, C.; Penel, C.; Dunand, C. Peroxidases have more functions than a Swiss army knife. Plant Cell Rep. 2005, 24, 255–265. [Google Scholar] [CrossRef]
- Kadota, Y.; Sklenar, J.; Derbyshire, P.; Stransfeld, L.; Asai, S.; Ntoukakis, V.; Jones, J.D.; Shirasu, K.; Menke, F.; Jones, A.; et al. Direct regulation of the NADPH oxidase RBOHD by the PRR-associated kinase BIK1 during plant immunity. Mol. Cell 2014, 54, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, M.A.; Lin, X.; Chan, T.-F.; Imtiaz, M.; Rehman, H.M.; Ali, M.A.; Baloch, F.S.; Atif, R.M.; Yang, S.H.; Chung, G. Characterization of Cellulose Synthase A (CESA) Gene Family in Eudicots. Biochem. Genet. 2019, 57, 248–272. [Google Scholar] [CrossRef] [PubMed]
- Malinovsky, F.G.; Fangel, J.U.; Willats, W.G. The role of the cell wall in plant immunity. Front. Plant Sci. 2014, 5, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinger, D.; Naumann, M.; Falter, C.; Zwikowics, C.; Jamrow, T.; Manisseri, C.; Somerville, S.C.; Voigt, C.A. Elevated early callose deposition results in complete penetration resistance to powdery mildew in Arabidopsis. Plant Physiol. 2013, 161, 1433–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacete, L.; Mélida, H.; Miedes, E.; Molina, A. Plant cell wall-mediated immunity: Cell wall changes trigger disease resistance responses. Plant J. 2018, 93, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, A.; Jedrzejczak-Rey, N.; Bednarek, P. Secondary metabolites in plant innate immunity: Conserved function of divergent chemicals. New Phytol. 2015, 206, 948–964. [Google Scholar] [CrossRef] [PubMed]
- Scheideler, M.; Schlaich, N.L.; Fellenberg, K.; Beissbarth, T.; Hauser, N.C.; Vingron, M.; Slusarenko, A.J.; Hoheisel, J.D. Monitoring the switch from housekeeping to pathogen defense metabolism in Arabidopsis thaliana using cDNA arrays. J. Biol. Chem. 2002, 277, 10555–10561. [Google Scholar] [CrossRef] [Green Version]
- Bolouri Moghaddam, M.R.; Van den Ende, W. Sugars and plant innate immunity. J. Exp. Bot. 2012, 63, 3989–3998. [Google Scholar] [CrossRef] [Green Version]
- Rojas, C.M.; Senthil-Kumar, M.; Tzin, V.; Mysore, K. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front. Plant Sci. 2014, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Kumar, Y.; Zhang, L.; Panigrahi, P.; Dholakia, B.B.; Dewangan, V.; Chavan, S.G.; Kunjir, S.M.; Wu, X.; Li, N.; Rajmohanan, P.R. Fusarium oxysporum mediates systems metabolic reprogramming of chickpea roots as revealed by a combination of proteomics and metabolomics. Plant Biotechnol. J. 2016, 14, 1589–1603. [Google Scholar] [CrossRef] [Green Version]
- Tavernier, V.; Cadiou, S.; Pageau, K.; Laugé, R.; Reisdorf-Cren, M.; Langin, T.; Masclaux-Daubresse, C. The plant nitrogen mobilization promoted by Colletotrichum lindemuthianum in Phaseolus leaves depends on fungus pathogenicity. J. Exp. Bot. 2007, 58, 3351–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulermo, T.; Bligny, R.; Gout, E.; Cotton, P. Amino acid changes during sunflower infection by the necrotrophic fungus B. cinerea. Plant Signal. Behav. 2009, 4, 859–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verslues, P.E.; Sharma, S. Proline metabolism and its implications for plant-environment interaction. Arab. Book Am. Soc. Plant Biol. 2010, 8, e0140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabro, G.; Kovács, I.; Pavet, V.; Szabados, L.; Alvarez, M.E. Proline accumulation and AtP5CS2 gene activation are induced by plant-pathogen incompatible interactions in Arabidopsis. Mol. Plant-Microbe Interact. 2004, 17, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Wu, Q.; He, T.; Lan, J.; Ding, L.; Liu, T.; Wu, Q.; Pan, Y.; Chen, T. Transcriptomic and Metabolomic Changes Triggered by Fusarium solani in Common Bean (Phaseolus vulgaris L.). Genes 2020, 11, 177. https://doi.org/10.3390/genes11020177
Chen L, Wu Q, He T, Lan J, Ding L, Liu T, Wu Q, Pan Y, Chen T. Transcriptomic and Metabolomic Changes Triggered by Fusarium solani in Common Bean (Phaseolus vulgaris L.). Genes. 2020; 11(2):177. https://doi.org/10.3390/genes11020177
Chicago/Turabian StyleChen, Limin, Quancong Wu, Tianjun He, Jianjun Lan, Li Ding, Tingfu Liu, Qianqian Wu, Yiming Pan, and Tingting Chen. 2020. "Transcriptomic and Metabolomic Changes Triggered by Fusarium solani in Common Bean (Phaseolus vulgaris L.)" Genes 11, no. 2: 177. https://doi.org/10.3390/genes11020177