Examination of Genomic and Transcriptomic Alterations in a Morphologically Stable Line, MU1, Generated by Intergeneric Pollination

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Genomic DNA Isolation and Whole-Genome Rresequencing
2.3. Distribution of Nucleotide Polymorphisms and Identification of Mutant Genes
2.4. RNA Isolation and Transcriptome Sequencing
2.5. RNA Extraction and Gene Expression Analysis by Quantitative PCR (qPCR)
3. Results
3.1. Phenotypic Characterization of MU1
3.2. Analysis of Genomic Variation in MU1
3.3. Classification of SNPs in MU1
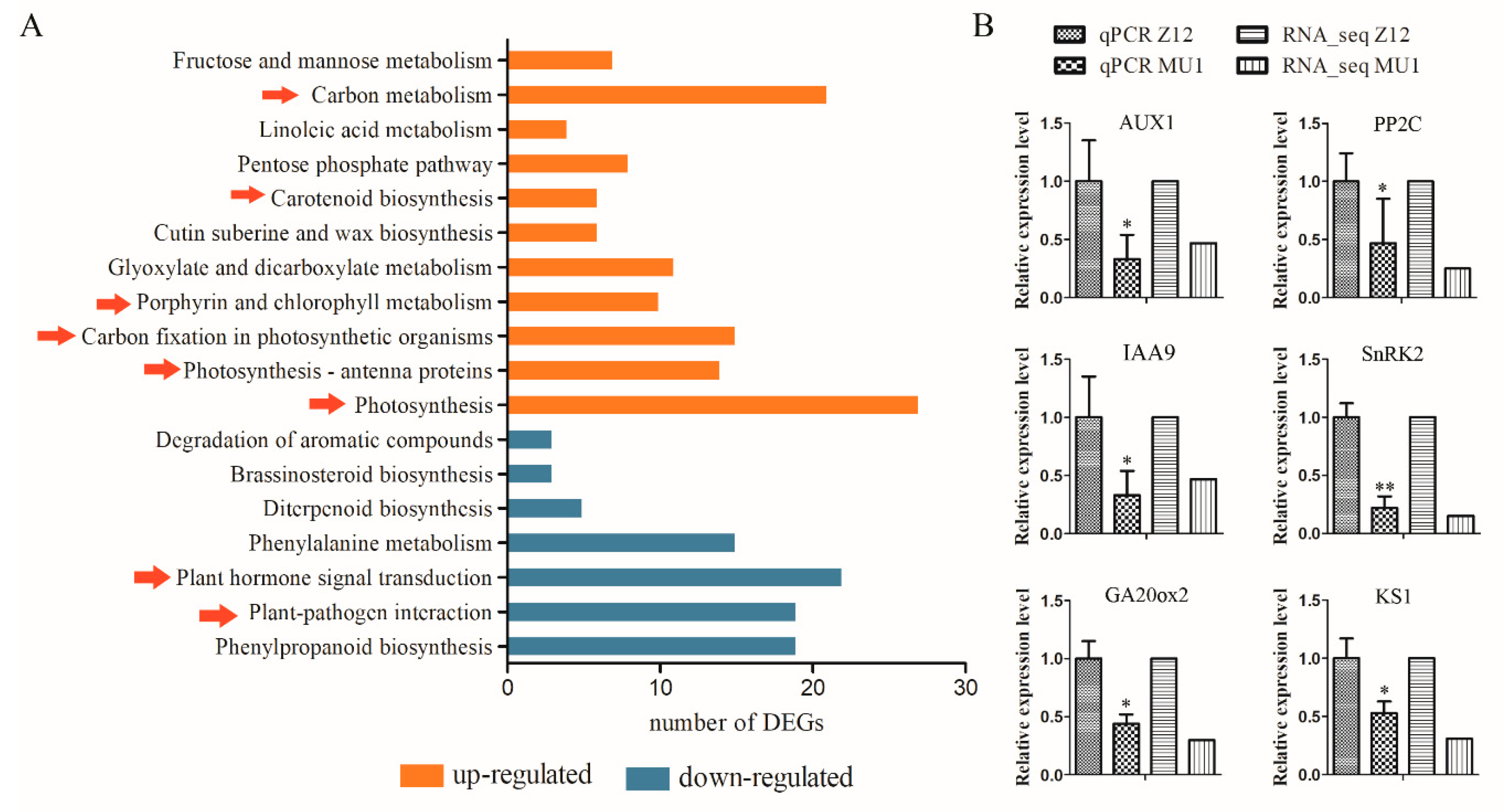
3.4. Changes in GO Annotations of Genes Differentially Expressed between MU1 and Z12
3.5. Changes in Signal Transduction Pathways in MU1
3.6. Verification of RNA-seq Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| qPCR | quantitative PCR |
| RILs | recombinant inbred lines |
| AUX | auxin transporter-like protein |
| PP2C | protein phosphatase 2C |
| GA | gibberellin acid |
| SnRK2 | SNF1-related kinase2 |
| DEG | differentially expressed genes |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Wang, H.; Chai, Y.; Chu, X.C.; Zhao, Y.Y.; Wu, Y.; Zhao, J.H.; Ngezahayo, F.; Xu, C.M.; Liu, B. Molecular characterization of a rice mutator-phenotype derived from an incompatible cross-pollination reveals transgenerational mobilization of multiple transposable elements and extensive epigenetic instability. BMC Plant Biol. 2009, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- McInnis, S.M.; Desikan, R.; Hancock, J.T.; Hiscock, S.J. Production of reactive oxygen species and reactive nitrogen species by angiosperm stigmas and pollen: Potential signalling crosstalk? New Phytol. 2006, 172, 221–228. [Google Scholar] [CrossRef]
- Stine, M.; Sears, B.B.; Keathley, D.E. Inheritance of plastids in interspecific hybrids of blue spruce and white spruce. Theor. Appl. Genet. 1989, 78, 768–774. [Google Scholar] [CrossRef]
- Dong, Z.Y.; Wang, Y.M.; Zhang, Z.J.; Shen, Y.; Lin, X.Y.; Ou, X.F.; Han, F.P.; Liu, B. Extent and pattern of DNA methylation alteration in rice lines derived from introgressive hybridization of rice and Zizania latifolia Griseb. Theor. Appl. Genet. 2006, 113, 196–205. [Google Scholar] [CrossRef]
- Labrador, M.; Farre, M.; Utzet, F.; Fontdevila, A. Interspecific hybridization increases transposition rates of Osvaldo. Mol. Biol. Evol. 1999, 16, 931–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wang, Y.; Shen, Y.; Guo, W.; Hao, S.; Liu, B. Extensive alterations in DNA methylation and transcription in rice caused by introgression from Zizania latifolia. Plant Mol. Biol. 2004, 54, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Dong, Z.Y.; Zhang, Z.J.; Lin, X.Y.; Shen, Y.; Zhou, D.; Liu, B. Extensive de Novo genomic variation in rice induced by introgression from wild rice (Zizania latifolia Griseb.). Genetics 2005, 170, 1945–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.H.; Zhang, D.; Bai, Y.; Zhang, Y.H.; Liu, Y.; Wu, Y.; Lin, X.Y.; Wen, J.W.; Xu, C.M.; Li, L.F.; et al. Genomewide variation in an introgression line of rice-Zizania revealed by whole-genome re-sequencing. PLoS ONE 2013, 8, e74479. [Google Scholar] [CrossRef]
- Sakai, S.; Kawakita, A.; Ooi, K.; Inoue, T. Variation in the strength of association among pollination systems and floral traits: Evolutionary changes in the floral traits of Bornean gingers (Zingiberaceae). Am. J. Bot. 2013, 100, 546–555. [Google Scholar] [CrossRef]
- Schiestl, F.P.; Johnson, S.D. Pollinator-mediated evolution of floral signals. Trends Ecol. Evol. 2013, 28, 307–315. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Genetic and epigenetic effects of plant-pathogen interactions: An evolutionary perspective. Mol. Plant 2011, 4, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holeski, L.M.; Jander, G.; Agrawal, A.A. Transgenerational defense induction and epigenetic inheritance in plants. Trends Ecol. Evol. 2012, 27, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Ellstrand, N.C. Does introgression of crop alleles into wild and weedy living populations create cryptic in situ germplasm banks? Mol. Ecol. 2018, 27, 38–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gressel, J. Dealing with transgene flow of crop protection traits from crops to their relatives. Pest. Manag. Sci. 2015, 71, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Nosil, P. Speciation with gene flow could be common. Mol. Ecol. 2008, 17, 2103–2106. [Google Scholar] [CrossRef]
- Rieseberg, L.H.; Willis, J.H. Plant speciation. Science 2007, 317, 910–914. [Google Scholar] [CrossRef]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Gaeta, R.T.; Pires, J.C.; Iniguez-Luy, F.; Leon, E.; Osborn, T.C. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 2007, 19, 3403–3417. [Google Scholar] [CrossRef] [Green Version]
- Madlung, A.; Masuelli, R.W.; Watson, B.; Reynolds, S.H.; Davison, J.; Comai, L. Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol. 2002, 129, 733–746. [Google Scholar] [CrossRef] [Green Version]
- Ozkan, H.; Levy, A.A.; Feldman, M. Allopolyploidy-induced rapid genome evolution in the wheat (Aegilops-Triticum) group. Plant Cell 2001, 13, 1735–1747. [Google Scholar] [PubMed] [Green Version]
- Salmon, A.; Ainouche, M.L.; Wendel, J.F. Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 2005, 14, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Shaked, H.; Kashkush, K.; Ozkan, H.; Feldman, M.; Levy, A.A. Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 2001, 13, 1749–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Fu, D.; Gong, H.; Qian, W.; Xia, W.; Pires, J.C.; Li, R.; Long, Y.; Mason, A.S.; Yang, T.J.; et al. De novo genetic variation associated with retrotransposon activation, genomic rearrangements and trait variation in a recombinant inbred line population of Brassica napus derived from interspecific hybridization with Brassica rapa. Plant J. 2011, 68, 212–224. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The significance of responses of the genome to challenge. Science 1984, 226, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Ribet, D.; Dewannieux, M.; Heidmann, T. An active murine transposon family pair: Retrotransposition of "master" MusD copies and ETn trans-mobilization. Genome Res. 2004, 14, 2261–2267. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, Z.L.; Li, X.W. Production of a highly asymmetric somatic hybrid between rice and Zizania latifolia (Griseb): Evidence for inter-genomic exchange. Theor. Appl. Genet. 1999, 98, 1099–1103. [Google Scholar] [CrossRef]
- Rubin, C.J.; Megens, H.J.; Barrio, A.M.; Maqbool, K.; Sayyab, S.; Schwochow, D.; Wang, C.; Carlborg, Ö.; Jern, P.; Jørgensen, C.B.; et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109, 19529–19536. [Google Scholar] [CrossRef] [Green Version]
- Rieseberg, L.H.; Raymond, O.; Rosenthal, D.M.; Lai, Z.; Livingstone, K.; Nakazato, T.; Durphy, J.L.; Schwarzbach, A.E.; Donovan, L.A.; Lexer, C. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 2003, 301, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Iskandar, H.M.; Simpson, R.S.; Casu, R.E.; Bonnett, G.D.; Maclean, D.J.; Manners, J.M. Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Mol. Biol. Rep. 2004, 22, 325–337. [Google Scholar] [CrossRef]
- Minoru, K.; Susumu, G. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- Depuydt, S.; Hardtke, C.S. Hormone signalling crosstalk in plant growth regulation. Curr. Biol. 2011, 21, R365–R373. [Google Scholar] [CrossRef]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Margis-Pinheiro, M.; Zhou, X.R.; Zhu, Q.H.; Dennis, E.S.; Upadhyaya, N.M. Isolation and characterization of a Ds-tagged rice (Oryza sativa L.) GA-responsive dwarf mutant defective in an early step of the gibberellin biosynthesis pathway. Plant Cell Rep. 2005, 23, 819–833. [Google Scholar] [CrossRef]
- Oikawa, T.; Koshioka, M.; Kojima, K.; Yoshida, H.; Kawata, M. A role of OsGA20ox1, encoding an isoform of gibberellin 20-oxidase, for regulation of plant stature in rice. Plant Mol. Biol. 2004, 55, 687–700. [Google Scholar] [CrossRef]
- Tezuka, D.; Ito, A.; Mitsuhashi, W.; Toyomasu, T.; Imai, R. The rice ent-KAURENE SYNTHASE LIKE 2 encodes a functional ent-beyerene synthase. Biochem. Biophys. Res. Commun. 2015, 460, 766–771. [Google Scholar] [CrossRef]
- Comai, L.; Tyagi, A.P.; Winter, K.; Holmes-Davis, R.; Reynolds, S.H.; Stevens, Y.; Byers, B. Phenotypic instability and rapid gene silencing in newly formed arabidopsis allotetraploids. Plant Cell 2000, 12, 1551–1568. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.H.; Wang, J.; Li, Z.Y. Different genome-specific chromosome stabilities in synthetic Brassica allohexaploids revealed by wide crosses with Orychophragmus. Ann. Bot. 2009, 104, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Hegarty, M.J.; Batstone, T.; Barker, G.L.; Edwards, K.J.; Abbott, R.J.; Hiscock, S.J. Nonadditive changes to cytosine methylation as a consequence of hybridization and genome duplication in Senecio (Asteraceae). Mol. Ecol. 2011, 20, 105–113. [Google Scholar] [CrossRef]
- Liu, B.; Vega, J.M.; Feldman, M. Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. II. Changes in low-copy coding DNA sequences. Genome 1998, 41, 535–542. [Google Scholar] [CrossRef]
- Malinska, H.; Tate, J.A.; Matyasek, R.; Leitch, A.R.; Soltis, D.E.; Soltis, P.S.; Kovarik, A. Similar patterns of rDNA evolution in synthetic and recently formed natural populations of Tragopogon (Asteraceae) allotetraploids. BMC Evol. Biol. 2010, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Petit, M.; Guidat, C.; Daniel, J.; Denis, E.; Montoriol, E.; Bui, Q.T.; Lim, K.Y.; Kovarik, A.; Leitch, A.R.; Grandbastien, M.A.; et al. Mobilization of retrotransposons in synthetic allotetraploid tobacco. New Phytol. 2010, 186, 135–147. [Google Scholar] [CrossRef]
- Cantu, D.; Vanzetti, L.S.; Sumner, A.; Dubcovsky, M.; Matvienko, M.; Distelfeld, A.; Michelmore, R.W.; Dubcovsky, J. Small RNAs, DNA methylation and transposable elements in wheat. BMC Genom. 2010, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Yu, H.J.; Mun, J.H.; Lee, S.C. Genome-wide discovery of DNA polymorphism in Brassica rapa. Mol. Genet. Genom. 2010, 283, 135–145. [Google Scholar] [CrossRef]
- Wang, X.W.; Zhao, Q.Y.; Luan, J.B.; Wang, Y.J.; Yan, G.H.; Liu, S.S. Analysis of a native whitefly transcriptome and its sequence divergence with two invasive whitefly species. BMC Genom. 2012, 13, 529. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2011, 30, 105–111. [Google Scholar] [CrossRef]
- Chen, J.Q.; Wu, Y.; Yang, H.; Bergelson, J.; Kreitman, M.; Tian, D. Variation in the ratio of nucleotide substitution and indel rates across genomes in mammals and bacteria. Mol. Biol. Evol. 2009, 26, 1523–1531. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.I.; Botstein, D.; Desai, M.M. Genetic variation and the fate of beneficial mutations in asexual populations. Genetics 2011, 188, 647–661. [Google Scholar] [CrossRef]
- Loewe, L.; Hill, W.G. The population genetics of mutations: Good, bad and indifferent. Philos. Trans. R Soc. Lond. B Biol. Sci. 2010, 365, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Akey, J.M.; Zhang, G.; Zhang, K.; Jin, L.; Shriver, M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome. Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef] [Green Version]
- Harris, G.P. Photosynthesis, productivity and growth. Adv. Limnol. 1978, 10, 1–171. [Google Scholar]
- Liu, Y.; Gu, D.D.; Ding, Y.F.; Wang, Q.S.; Li, G.H.; Wang, S.H. The relationship between nitrogen, auxin and cytokinin in the growth regulation of rice (Oryza sativa L.) tiller buds. Aust. J. Crop Sci. 2011, 5, 1019–1026. [Google Scholar]
- Lo, S.F.; Yang, S.Y.; Chen, K.T.; Hsing, Y.L.; Zeevaart, J.A.D.; Chen, L.J.; Yu, S.M. A Novel Class of Gibberellin 2-Oxidases Control Semidwarfism, Tillering, and Root Development in Rice. Plant Cell 2008, 20, 2603–2618. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, T.; Miura, K.; Itoh, H.; Tatsumi, T.; Ueguchi-Tanaka, M.; Ishiyama, K.; Kobayashi, M.; Agrawal, G.K.; Takeda, S.; Abe, K.; et al. An overview of gibberellin metabolism enzyme genes and their related mutants in rice. Plant Physiol. 2004, 134, 1642–1653. [Google Scholar] [CrossRef] [Green Version]
- Ashikari, M.; Sakakibara, H.; Lin, S.Y.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Sano, H.; Seo, S.; Orudgev, E.; Youssefian, S.; Ishizuka, K. Expression of the gene for a small GTP binding protein in transgenic tobacco elevates endogenous cytokinin levels, abnormally induces salicylic acid in response to wounding, and increases resistance to tobacco mosaic virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 10556–10560. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Line | Plant Height (cm) | Germination Rate (%) | No. of Effective Panicles | 1000-Grain Weight (g) | No. of Grains Per Ear | Yield Per Acre (kg) |
|---|---|---|---|---|---|---|
| MU1 | 82.4 ± 6.8 ** | 81.2 ± 4.6 * | 22.2 ± 3.8 ** | 22.22 ± 0.43 * | 103.5 ± 6.4 ** | 560.63 ± 6.86 ** |
| Z12 | 95.9 ± 2.2 | 86.3 ± 1.7 | 16.4 ± 2.4 | 21.35 ± 0.46 | 142.7 ± 5.6 | 532.42 ± 5.17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, W.-L.; Zhao, M.-J.; Yang, X.-B.; Zhang, A.-X.; Wang, N.-N.; Xu, Z.-S.; Ma, J. Examination of Genomic and Transcriptomic Alterations in a Morphologically Stable Line, MU1, Generated by Intergeneric Pollination. Genes 2020, 11, 199. https://doi.org/10.3390/genes11020199
Meng W-L, Zhao M-J, Yang X-B, Zhang A-X, Wang N-N, Xu Z-S, Ma J. Examination of Genomic and Transcriptomic Alterations in a Morphologically Stable Line, MU1, Generated by Intergeneric Pollination. Genes. 2020; 11(2):199. https://doi.org/10.3390/genes11020199
Chicago/Turabian StyleMeng, Wei-Long, Meng-Jie Zhao, Xiang-Bo Yang, An-Xing Zhang, Ning-Ning Wang, Zhao-Shi Xu, and Jian Ma. 2020. "Examination of Genomic and Transcriptomic Alterations in a Morphologically Stable Line, MU1, Generated by Intergeneric Pollination" Genes 11, no. 2: 199. https://doi.org/10.3390/genes11020199