Analysis of Long Non-Coding RNAs and mRNAs Associated with Lactation in the Crop of Pigeons (Columba livia)

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. Total RNA Extraction
2.4. Library Preparation for lncRNA Sequencing
2.5. Quality Control, Mapping, and Assembly of Transcriptomic Data
2.6. Identification of lncRNAs and Target Gene Prediction
2.7. Differential Expression and Functional Enrichment Analysis
2.8. qRT-PCR Assay
3. Results
3.1. Overview of RNA Sequencing
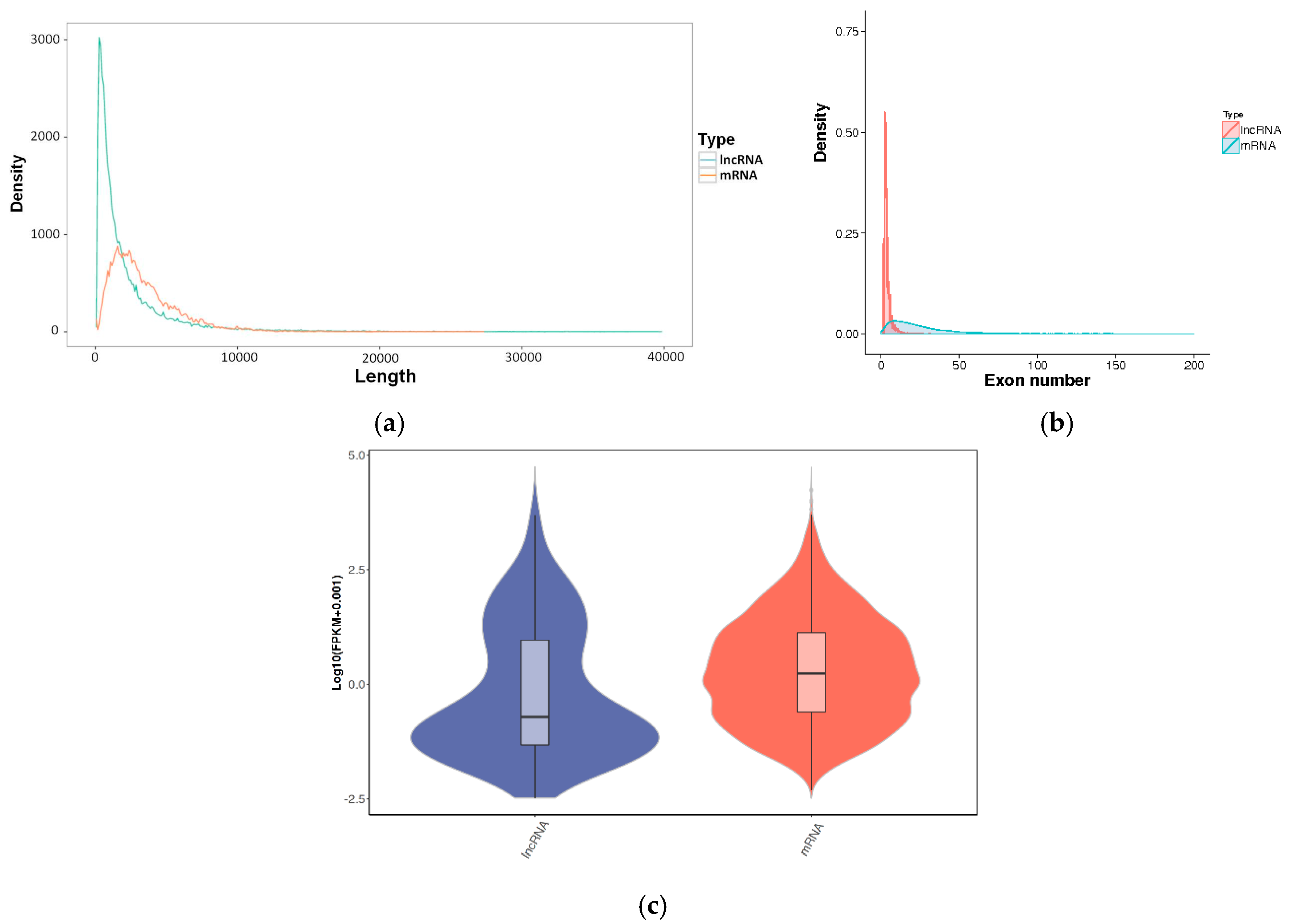
3.2. Identification and Characterization of lncRNAs
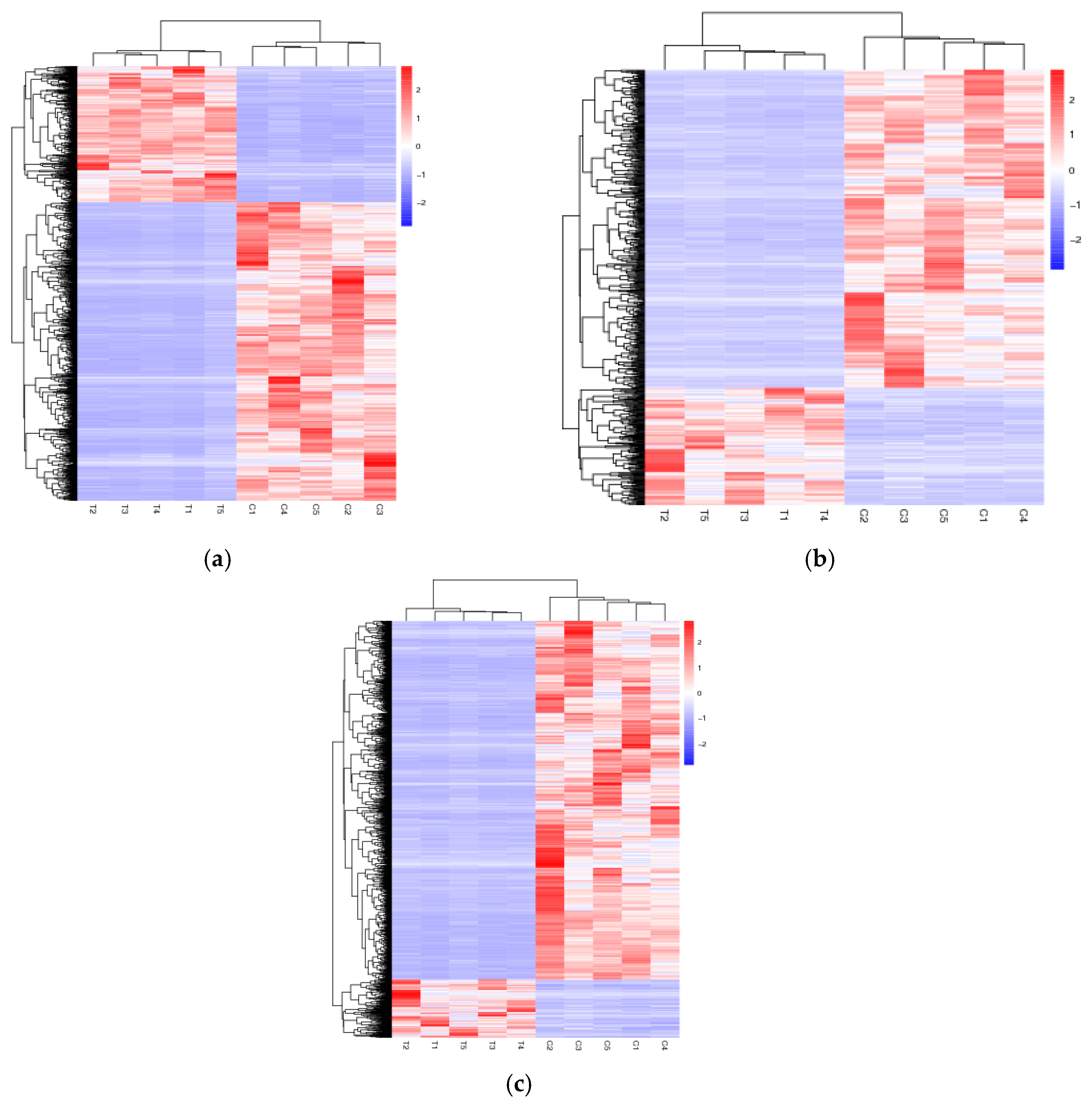
3.3. Differential Expression Analysis
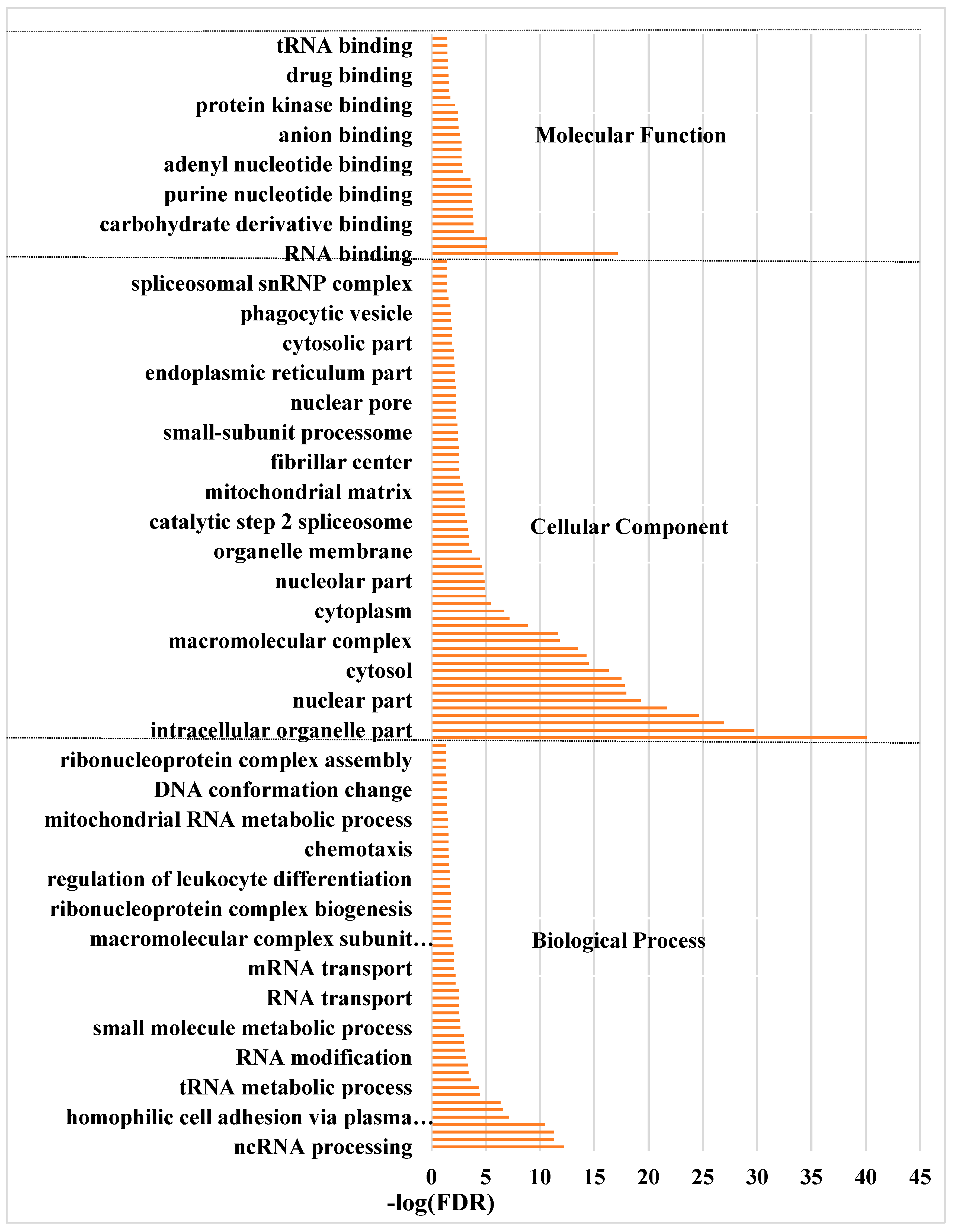
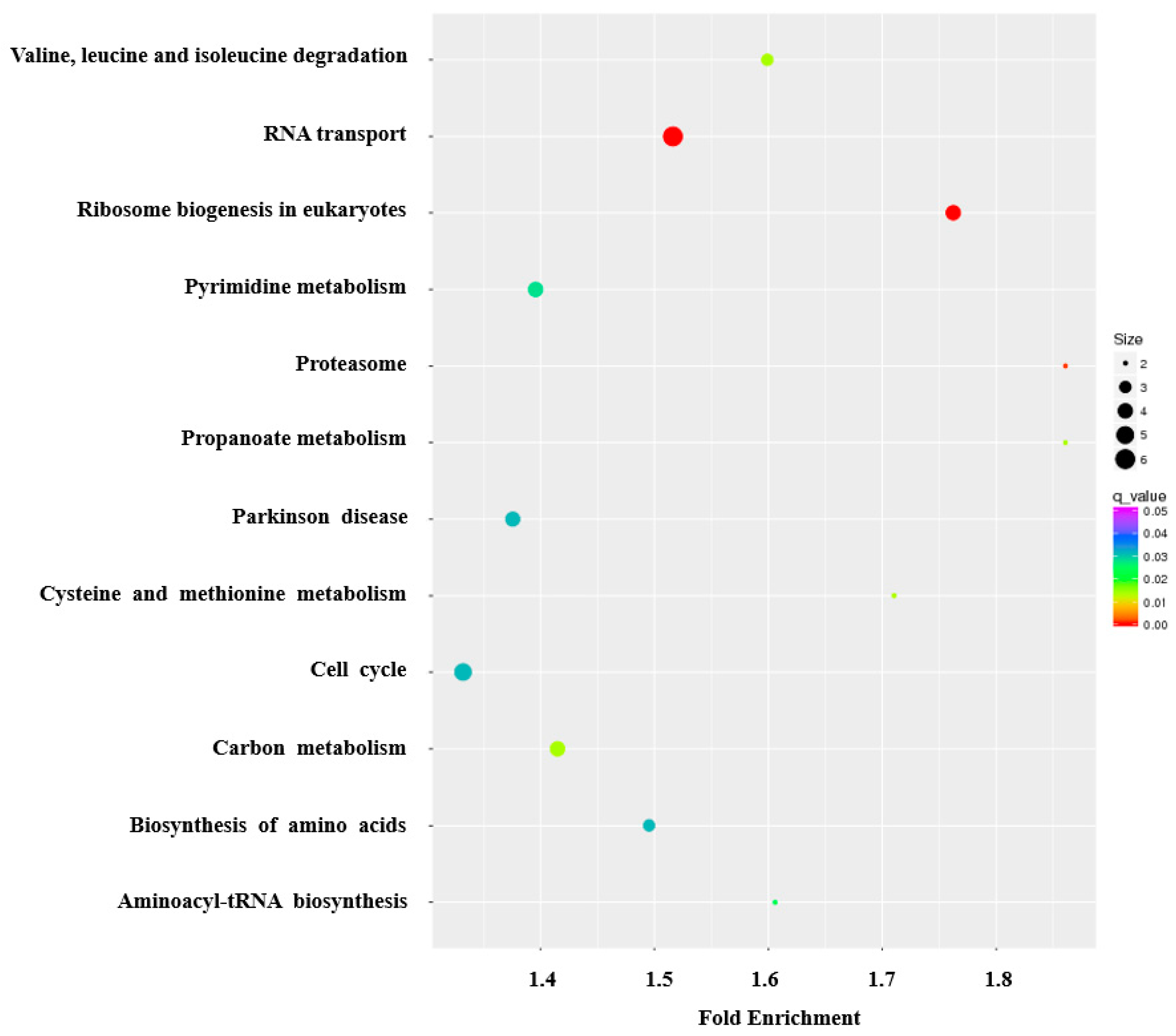
3.4. Function Enrichment Related to Pigeon Milk Production
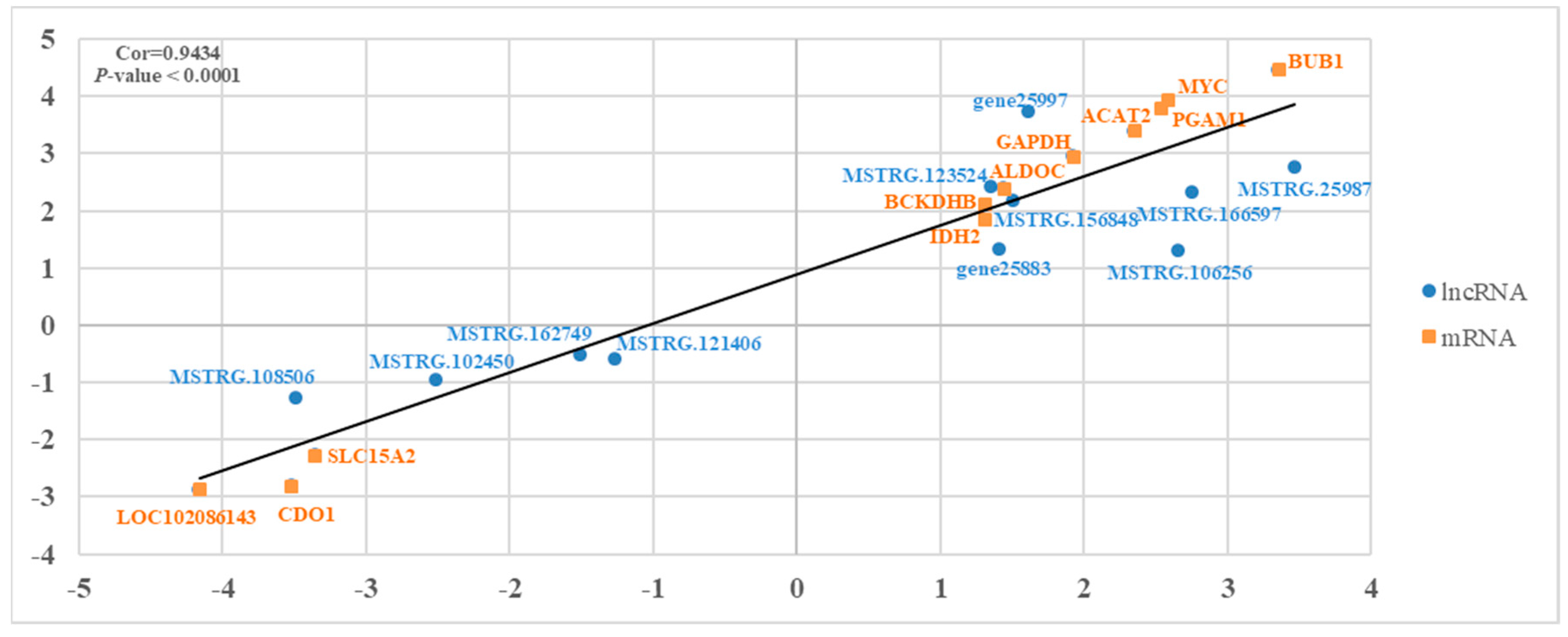
3.5. qRT-PCR Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shapiro, M.D.; Kronenberg, Z.; Li, C.; Domyan, E.T.; Pan, H.; Campbell, M.; Tan, H.; Huff, C.D.; Hu, H.; Vickrey, A.I.; et al. Genomic diversity and evolution of the head crest in the rock pigeon. Science 2013, 339, 1063–1067. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, M.J.; Haring, V.R.; McColl, K.A.; Monaghan, P.; Donald, J.A.; Nicholas, K.R.; Moore, R.J.; Crowley, T.M. Histological and global gene expression analysis of the ‘lactating’ pigeon crop. BMC Genomics 2011, 12, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.; Owen, R. Observations on Certain Parts of the Animal Economy: Inclusive of Several Papers from the Philosophical Transactions, etc; Haswell, Barrington, and Haswell: New Orleans, LA, USA, 1840. [Google Scholar]
- JaVV, P. Histologie de la secretion oesophagienne du manchot empereur. In Proceedings of the 13th International Ornithological Congress, Ithaca, NY, USA, 17–24 June 1962; Volume 2, pp. 1085–1094. [Google Scholar]
- Studer-Thiersch, A. Beitrage zur brutbiologie der flamingos (Gattung phoenicopterus). Zool. Gart. 1967, 34, 159–229. [Google Scholar]
- Pukac, L.A.; Horseman, N.D. Regulation of cloned prolactin-inducible genes in pigeon crop. Mol. Endocrinol. 1987, 1, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horseman, N.D.; Buntin, J.D. Regulation of pigeon cropmilk secretion and parental behaviors by prolactin. Annu. Rev. Nutr. 1995, 15, 213–238. [Google Scholar] [CrossRef]
- Gillespie, M.J.; Crowley, T.M.; Haring, V.R.; Wilson, S.L.; Harper, J.A.; Payne, J.S.; Green, D.; Monaghan, P.; Stanley, D.; Donald, J.A.; et al. Transcriptome analysis of pigeon milk production—Role of cornification and triglyceride synthesis genes. BMC Genomics 2013, 14, 169. [Google Scholar] [CrossRef] [Green Version]
- Damas, J.; O’Connor, R.; Farré, M.; Lenis, V.P.E.; Martell, H.J.; Mandawala, A.; Fowler, K.; Joseph, S.; Swain, M.T.; Griffin, D.K.; et al. Upgrading short-read animal genome assemblies to chromosome level using comparative genomics and a universal probe set. Genome Res. 2017, 27, 875–884. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.; Campbell, M.; Keays, D.A.; Edelman, N.; Kapusta, A.; Maclary, E.; Doyman, E.T.; Suh, A.; Warren, W.C.; Yandell, M.; et al. Improved Genome Assembly and Annotation for the Rock Pigeon (Columba livia). G3 Genes Genomes Genet. 2018, 8, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, H.; Matsuda, Y.; Yamamoto, M.; Kamiya, S.; Ishiwata, T. Expression and role of long non-coding RNA H19 in carcinogenesis. Front. Biosci. 2018, 23, 614–625. [Google Scholar] [CrossRef]
- Bernier-Dodier, P.; Girard, C.L.; Talbot, B.G.; Lacasse, P. Effect of dry period management on mammary gland function and its endocrine regulation in dairy cows. J. Dairy Sci. 2011, 94, 4922–4936. [Google Scholar] [CrossRef] [Green Version]
- Broccolo, F.; Maran, V.; Oggioni, M.; Matteoli, B.; Greppi, G.; Ceccherini-Nelli, L.; Fusetti, L. Development and validation of a dedicated microarray for the evaluation of bovine mammary gland health status and milk quality. Mol. Biotechnol. 2013, 54, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Seeth, M.; Hoedemaker, M.; Kromker, V. Physiological processes in the mammary gland tissue of dairy cows during the dry period. Berl. Munch. Tierarztl. Wochenschr. 2015, 128, 76–83. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nature Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Database for Annotation, Visualization and Integrated Discovery (DAVID). Available online: https://david.ncifcrf.gov/ (accessed on 7 February 2020).
- Horseman, N.D. A prolactin-inducible gene product which is a member of the calpactin/lipocortin family. Mol. Endocrinol. 1989, 3, 773–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, M.J.; Stanley, D.; Chen, H.; Donald, J.A.; Nicholas, K.R.; Moore, R.J.; Crowley, T.M. Functional similarities between pigeon ‘milk’ and mammalian milk: Induction of immune gene expression and modification of the microbiota. PLoS ONE 2012, 7, e48363. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.R.; Jury, N.J.; Gregerson, K.A.; Horseman, N.D.; Hernandez, L.L. Characterization of mammary-specific disruptions for Tph1 and Lrp5 during murine lactation. Sci. Rep. 2017, 7, 15155. [Google Scholar] [CrossRef] [PubMed]
- Cases, S.; Stone, S.J.; Zhou, P.; Yen, E.; Tow, B.; Lardizabal, K.D.; Voelker, T.; Farese, R.V., Jr. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Boil. Chem. 2001, 276, 38870–38876. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.; Widjaja-Adhi, M.A.; Palczewski, G.; von Lintig, J. Transport of vitamin A across blood-tissue barriers is facilitated by STRA6. FASEB J. 2016, 30, 2985–2995. [Google Scholar] [CrossRef] [Green Version]
- Talhouk, R.S.; Elble, R.C.; Bassam, R.; Daher, M.; Sfeir, A.; Mosleh, L.A.; El-Khoury, H.; Hamoui, S.; Pauli, B.U.; El-Sabban, M.E. Developmental expression patterns and regulation of connexins in the mouse mammary gland: Expression of connexin30 in lactogenesis. Cell Tissue Res. 2005, 319, 49–59. [Google Scholar] [CrossRef]
- Wang, W.; Xue, W.; Jin, B.; Zhang, X.; Ma, F.; Xu, X. Candidate gene expression affects intramuscular fat content and fatty acid composition in pigs. J. Appl. Genet. 2013, 54, 113–118. [Google Scholar] [CrossRef]
- He, Y.; Ding, Y.; Zhan, F.; Zhang, H.; Han, B.; Hu, G.; Zhao, K.; Yang, N.; Yu, Y.; Mao, L.; et al. The conservation and signatures of lincRNAs in Marek’s disease of chicken. Sci. Rep. 2015, 5, 15184. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lin, Y.; Wu, J. Long non-coding RNA expression profiling of mouse testis during postnatal development. PLoS ONE 2013, 8, e75750. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Wu, Y.; Yang, Y.; Yang, Y.T.; Wang, Z.; Yuan, J.; Yang, Y.; Hua, C.; Fan, X.; Niu, G.; et al. Comprehensive analysis of long non-coding RNAs highlights their spatio-temporal expression patterns and evolutional conservation in Sus scrofa. Sci. Rep. 2017, 7, 43166. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Adelson, D.L. Bovine ncRNAs are abundant, primarily intergenic, conserved and associated with regulatory genes. PLoS ONE 2012, 7, e42638. [Google Scholar] [CrossRef] [Green Version]
- Koufariotis, L.T.; Chen, Y.P.; Chamberlain, A.; Vander Jagt, C.; Hayes, B.J. A catalogue of novel bovine long noncoding RNA across 18 tissues. PLoS ONE 2015, 10, e0141225. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhao, Y.; Lai, F.; Chu, M.; Hao, Y.; Feng, Y.; Zhang, H.; Liu, J.; Cheng, M.; Li, L.; et al. LncRNA as ceRNAs may be involved in lactation process. Oncotarget 2017, 8, 98014–98028. [Google Scholar] [CrossRef] [Green Version]
- Shore, A.N.; Rosen, J.M. Regulation of mammary epithelial cell homeostasis by lncRNAs. Int. J. Biochem. Cell Biol. 2014, 54, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.Z.; Shi, K.; Wu, X.H.; Xue, M.Y.; Wei, Z.H.; Liu, J.X.; Liu, H.Y. Lactation-related metabolic mechanism investigated based on mammary gland metabolomics and 4 biofluids’ metabolomics relationships in dairy cows. BMC Genomics 2017, 18, 936. [Google Scholar] [CrossRef] [Green Version]
- Davies, C.R.; Morris, J.S.; Griffiths, M.R.; Page, M.J.; Pitt, A.; Stein, T.; Gusterson, B.A. Proteomic analysis of the mouse mammary gland is a powerful tool to identify novel proteins that are differentially expressed during mammary development. Proteomics 2006, 6, 5694–5704. [Google Scholar] [CrossRef]
- Zheng, X.; Ning, C.; Zhao, P.; Feng, W.; Jin, Y.; Zhou, L.; Yu, Y.; Liu, J. Integrated analysis of long noncoding RNA and mRNA expression profiles reveals the potential role of long noncoding RNA in different bovine lactation stages. J. Dairy Sci. 2018, 101, 11061–11073. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Lee, S.B.; Hwang, J.H.; Lim, J.N.; Jung, U.S.; Kim, M.J.; Kang, H.S.; Choi, S.H.; Lee, J.S.; Roh, S.G.; et al. Proteomic Analysis Reveals PGAM1 Altering cis-9, trans-11 Conjugated Linoleic Acid Synthesis in Bovine Mammary Gland. Lipids 2015, 50, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Fuke, G.; Nornberg, J.L. Systematic evaluation on the effectiveness of conjugated linoleic acid in human health. Crit. Rev. Food Sci. Nutr. 2017, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, L.A.; Sadri, H.; von Soosten, D.; Danicke, S.; Egert, S.; Stehle, P.; Sauerwein, H. Changes in tissue abundance and activity of enzymes related to branched-chain amino acid catabolism in dairy cows during early lactation. J. Dairy Sci. 2019, 102, 3556–3568. [Google Scholar] [CrossRef] [Green Version]
- Goudarzi, A. The recent insights into the function of ACAT1: A possible anti-cancer therapeutic target. Life Sci. 2019, 232, 116592. [Google Scholar] [CrossRef]
- Chu, M.; Zhao, Y.; Yu, S.; Hao, Y.; Zhang, P.; Feng, Y.; Zhang, H.; Ma, D.; Liu, J.; Cheng, M.; et al. miR-15b negatively correlates with lipid metabolism in mammary epithelial cells. Am. J. Physiol. Cell Physiol. 2018, 314, C43–C52. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Ji, H.M.; Park, Y.; Sun, K. Flow maximization analysis of cell cycle pathway activation status in breast cancer subtypes. In Proceedings of the IEEE International Conference on Big Data & Smart Computing, Jeju, Korea, 13–16 February 2017. [Google Scholar]
- Heike, G.; Shinsuke, T.; Parsons, W.J.; Natalja, P.; Alfred, B.; Helmut Erich, G.; Wolfram, M. Overexpression of the mitotic checkpoint genes BUB1, BUBR1, and BUB3 in gastric cancer—Association with tumour cell proliferation. J. Pathol. 2003, 200, 16–22. [Google Scholar]
- He, Z.; Kannan, N.; Nemirovsky, O.; Chen, H.; Connell, M.; Taylor, B.; Jiang, J.; Pilarski, L.M.; Fleisch, M.C.; Niederacher, D. BRCA1 controls the cell division axis and governs ploidy and phenotype in human mammary cells. Oncotarget 2015, 8, 32461–32475. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Raw Bases | Clean Bases | Clean Reads Rate (%) | Clean Q30 Rate (%) | Mapped Reads | Mapping Rate (%) | MultiMap Reads | MultiMap Rate (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| T1 | 130,167,700 | 118,548,640 | 19,525,155,000 | 17,782,296,000 | 91.07 | 93.46 | 102,594,735 | 86.54 | 1,762,257 | 1.49 |
| T2 | 126,086,574 | 115,719,768 | 18,912,986,100 | 17,357,965,200 | 91.78 | 93.67 | 100,909,904 | 87.20 | 1,870,468 | 1.62 |
| T3 | 129,766,416 | 118,451,046 | 19,464,962,400 | 17,767,656,900 | 91.28 | 93.18 | 100,393,163 | 84.75 | 2,107,619 | 1.78 |
| T4 | 131,819,754 | 117,900,806 | 19,772,963,100 | 17,685,120,900 | 89.44 | 92.68 | 100,695,085 | 85.41 | 1,779,623 | 1.51 |
| T5 | 134,001,318 | 119,376,642 | 20,100,197,700 | 17,906,496,300 | 89.09 | 93.31 | 101,578,639 | 85.09 | 1,756,397 | 1.47 |
| C1 | 123,105,520 | 109,577,762 | 18,465,828,000 | 16,436,664,300 | 89.01 | 93.49 | 95,044,586 | 86.74 | 1,440,056 | 1.31 |
| C2 | 128,175,504 | 119,045,316 | 19,226,325,600 | 17,856,797,400 | 92.88 | 93.80 | 106,685,232 | 89.62 | 1,338,762 | 1.12 |
| C3 | 131,962,046 | 120,026,894 | 19,794,306,900 | 18,004,034,100 | 90.96 | 93.37 | 103,784,877 | 86.47 | 1,380,399 | 1.15 |
| C4 | 125,346,964 | 111,498,914 | 18,802,044,600 | 16,724,837,100 | 88.95 | 93.55 | 95,706,661 | 85.84 | 1,420,326 | 1.27 |
| C5 | 131,369,282 | 118,473,848 | 19,705,392,300 | 17,771,077,200 | 90.18 | 93.45 | 103,127,477 | 87.05 | 1,496,137 | 1.26 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, H.; Ni, A.; Ge, P.; Li, Y.; Shi, L.; Wang, P.; Fan, J.; Isa, A.M.; Sun, Y.; Chen, J. Analysis of Long Non-Coding RNAs and mRNAs Associated with Lactation in the Crop of Pigeons (Columba livia). Genes 2020, 11, 201. https://doi.org/10.3390/genes11020201
Ma H, Ni A, Ge P, Li Y, Shi L, Wang P, Fan J, Isa AM, Sun Y, Chen J. Analysis of Long Non-Coding RNAs and mRNAs Associated with Lactation in the Crop of Pigeons (Columba livia). Genes. 2020; 11(2):201. https://doi.org/10.3390/genes11020201
Chicago/Turabian StyleMa, Hui, Aixin Ni, Pingzhuang Ge, Yunlei Li, Lei Shi, Panlin Wang, Jing Fan, Adamu Mani Isa, Yanyan Sun, and Jilan Chen. 2020. "Analysis of Long Non-Coding RNAs and mRNAs Associated with Lactation in the Crop of Pigeons (Columba livia)" Genes 11, no. 2: 201. https://doi.org/10.3390/genes11020201
APA StyleMa, H., Ni, A., Ge, P., Li, Y., Shi, L., Wang, P., Fan, J., Isa, A. M., Sun, Y., & Chen, J. (2020). Analysis of Long Non-Coding RNAs and mRNAs Associated with Lactation in the Crop of Pigeons (Columba livia). Genes, 11(2), 201. https://doi.org/10.3390/genes11020201