Genome-Wide Identification of lncRNAs During Rice Seed Development

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
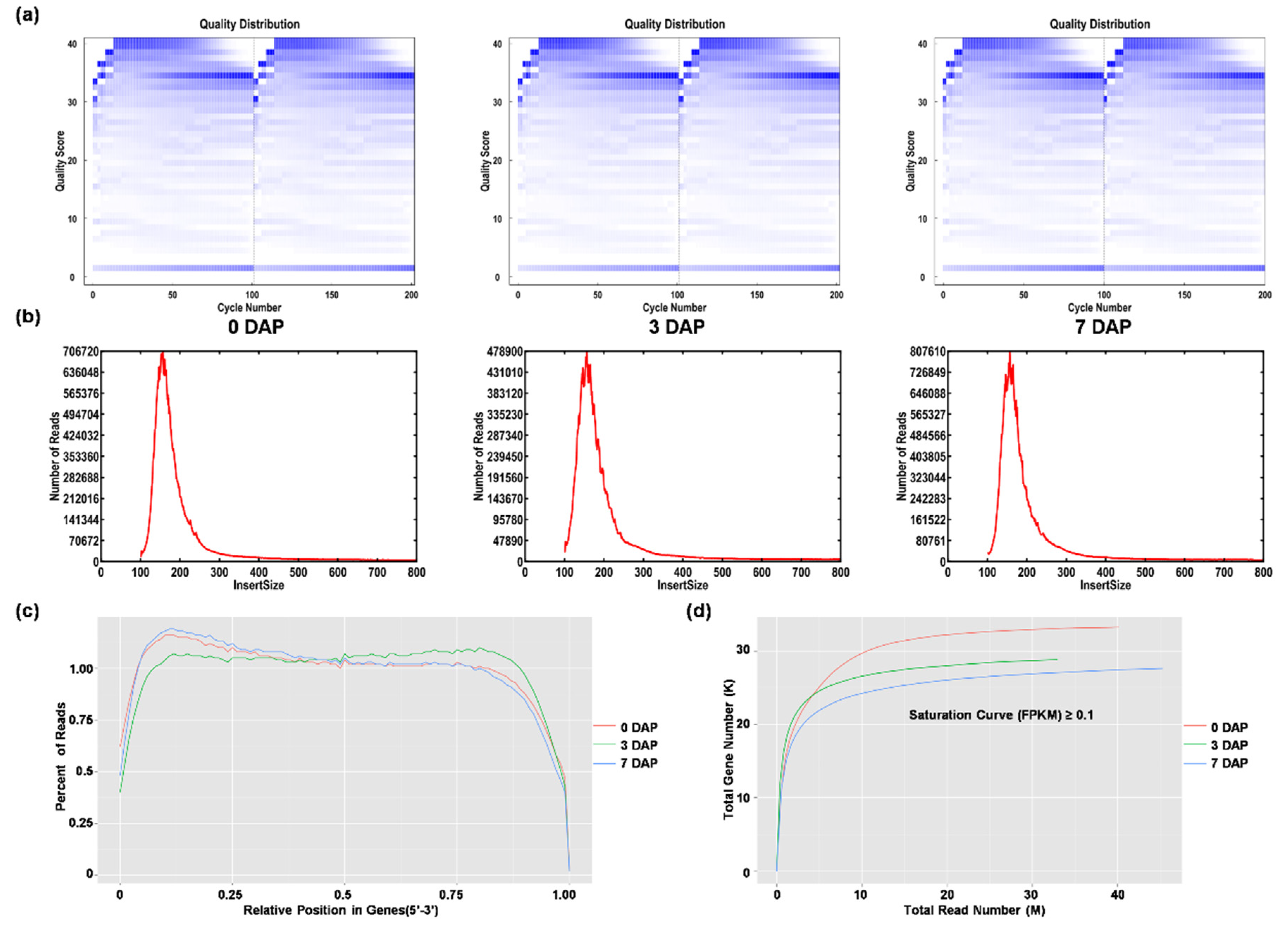
2.2. Paired-End RNA Sequencing
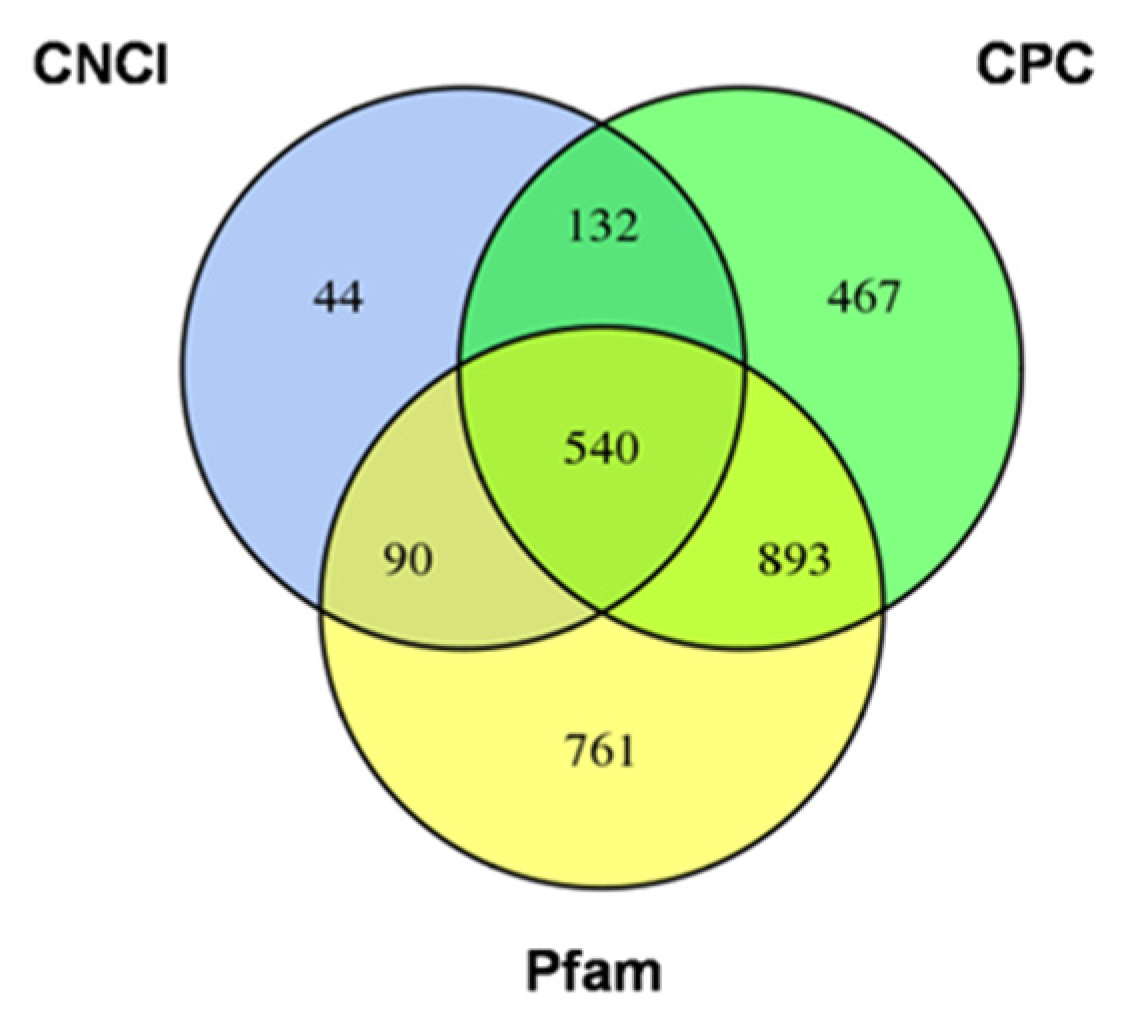
2.3. The Long Non-Coding RNAs Prediction
2.4. Bioinformatic Analysis
2.5. RNA Extraction and Reverse Transcription
2.6. Semi-Quantitative PCR and qRT-PCR Analysis
2.7. RNA Interference Vector Construction, Plant Transformation and Phenotype Characterization
3. Results
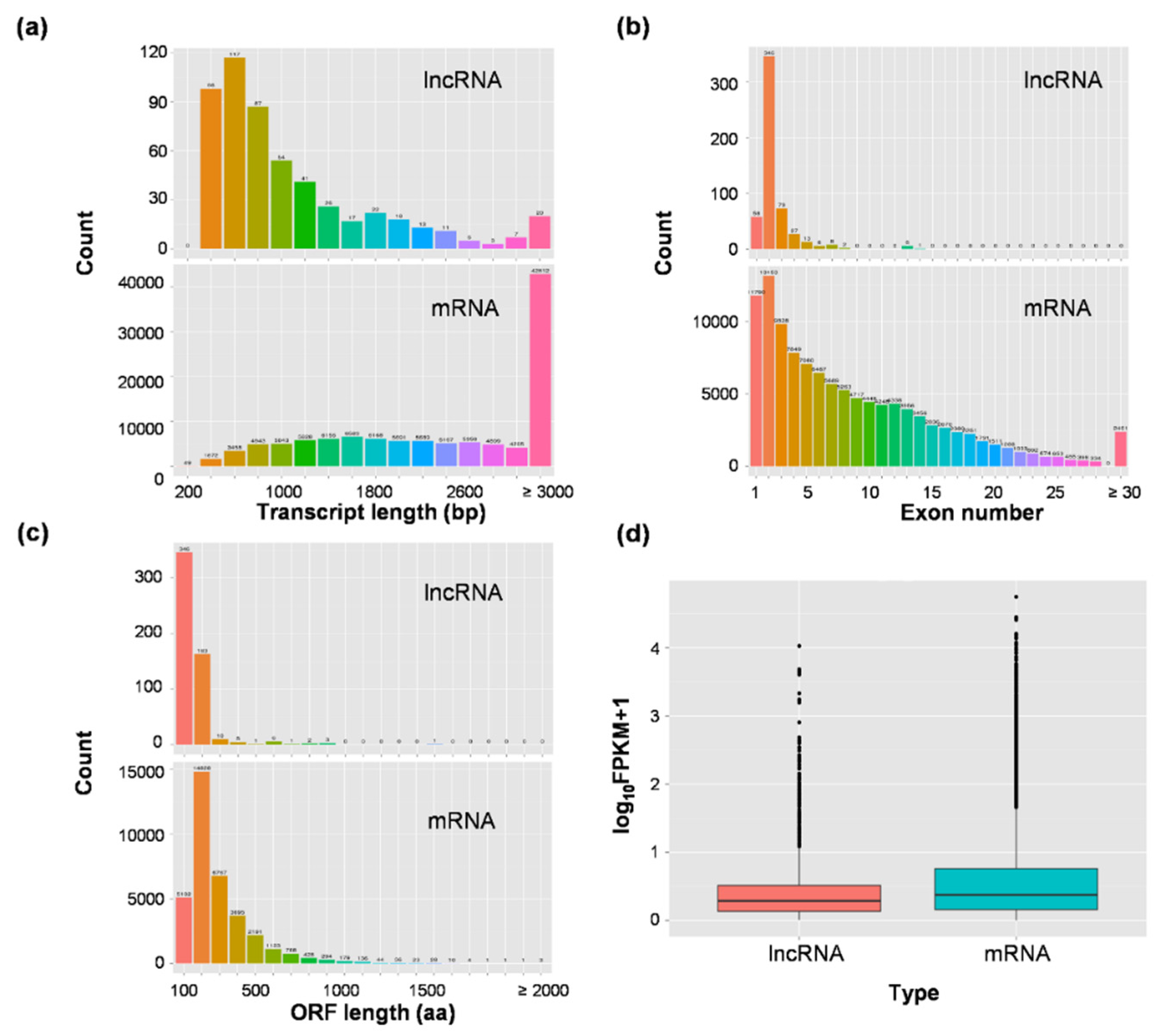
3.1. Genome-Wide Identification of Long Non-Coding RNAs in Rice Developing Seeds
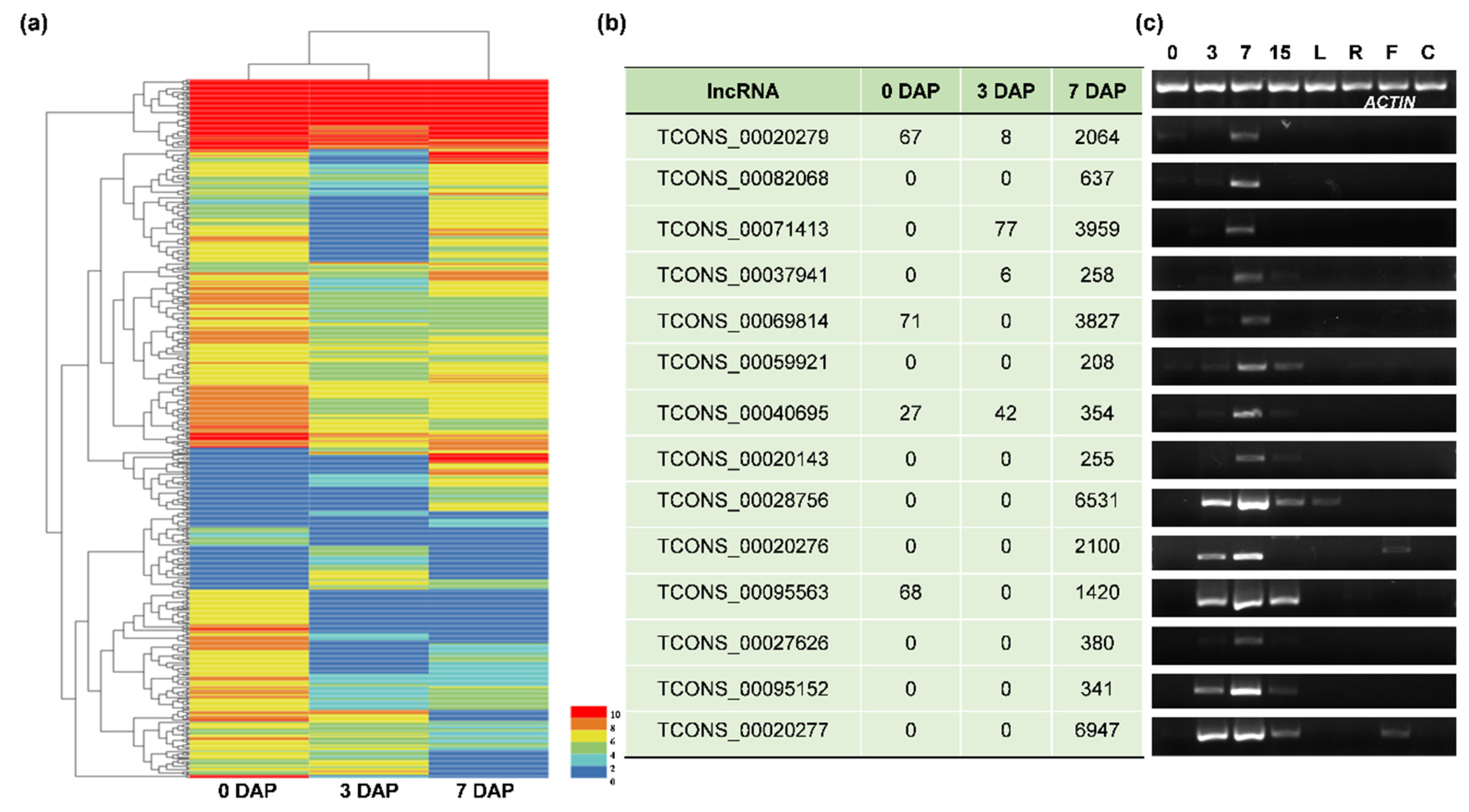
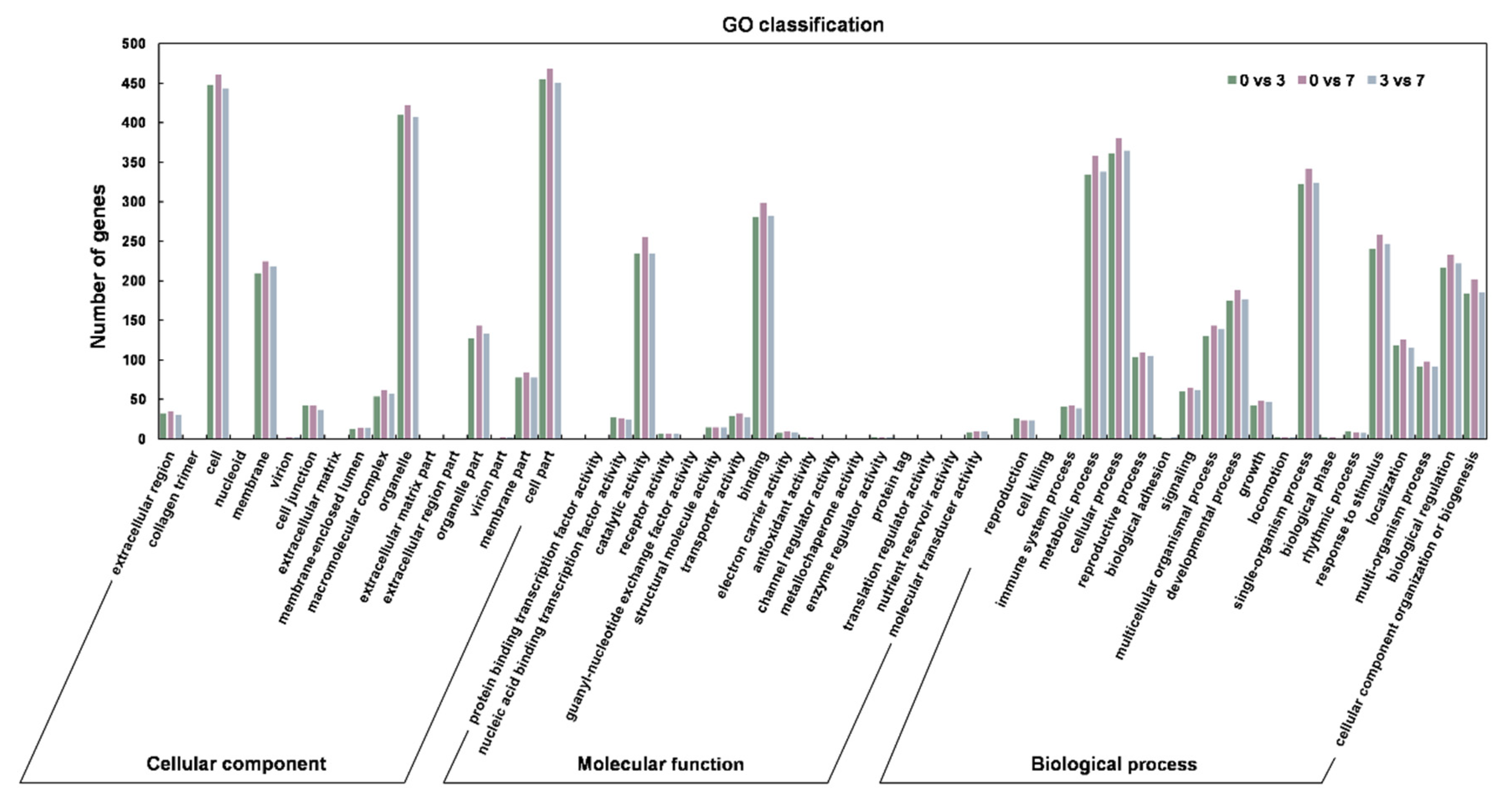
3.2. The Identification and Verification of Differentially Expressed lncRNAs (DELncs)
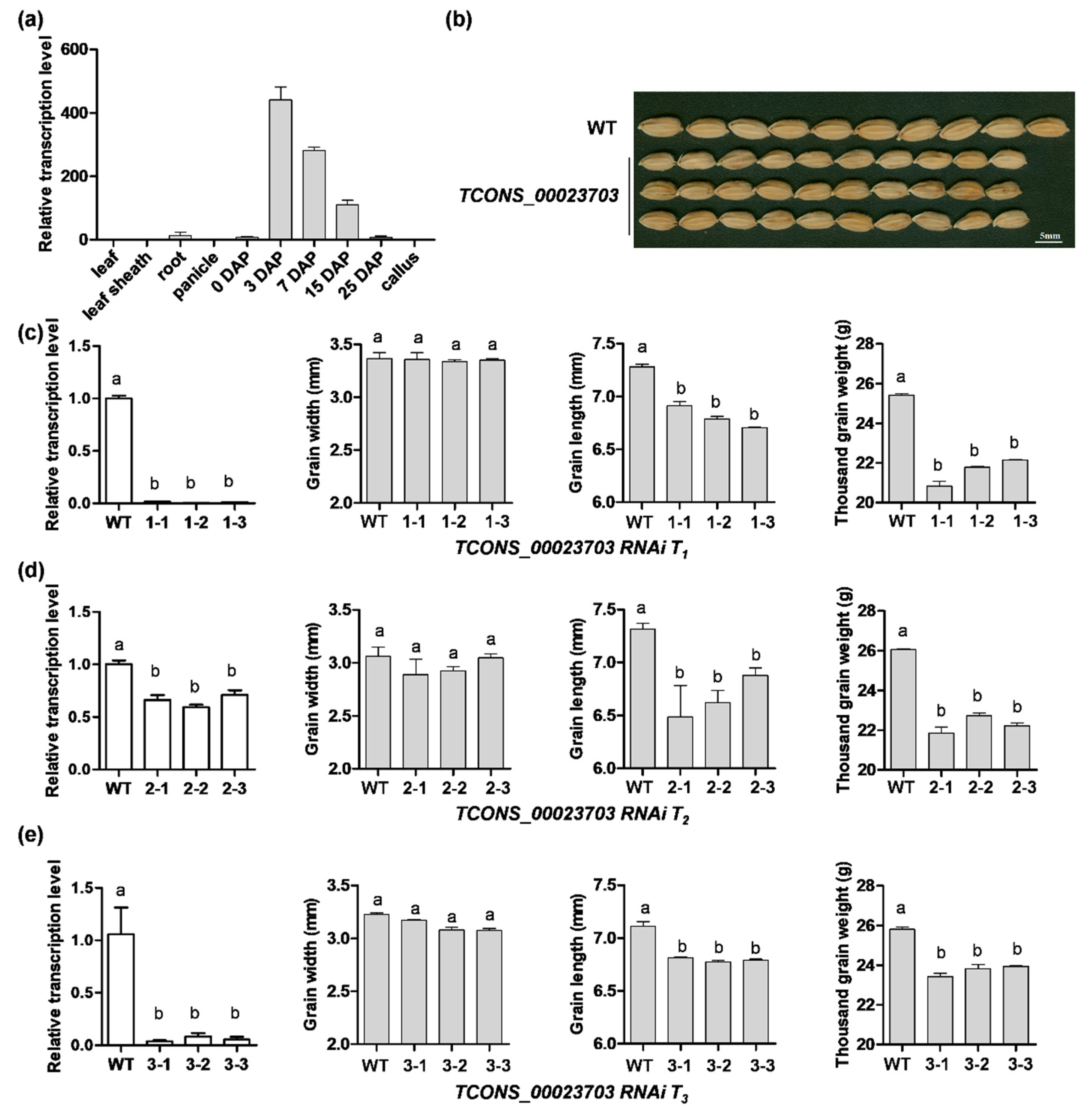
3.3. RNA Interference Analysis Reveals the lncRNA Participating in Seed Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [PubMed] [Green Version]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Mujahid, H.; Hou, Y.; Nallamilli, B.R.; Peng, Z. Plant long non-coding RNA: A new frontier of gene regulation. Am. J. Plant Sci. 2013, 4, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Ricciuti, B.; Mencaroni, C.; Paglialunga, L. Long noncoding RNAs: New insights into non-small cell lung cancer biology, diagnosis and therapy. Med. Oncol. 2016, 33, 18. [Google Scholar] [CrossRef]
- Mishra, A.; Bohra, A. Non-coding RNAs and plant male sterility: Current knowledge and future prospects. Plant Cell Rep. 2018, 37, 177–191. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, P.; Wang, L. Long non-coding RNA HOTAIR in carcinogenesis and metastasis. Acta Biochimica Et Biophysica Sinica 2014, 46, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Shafiq, S.; Li, J.; Sun, Q. Functions of plants long non-coding RNAs. Biochimica Et Biophysica Acta 2016, 1859, 155–162. [Google Scholar] [CrossRef]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar] [CrossRef]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.C.; Kean, M.J.; Xu, J.; Chua, N.H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Niu, Q.W.; Wu, H.W.; Liu, J.; Ye, J.; Yu, N.; Chua, N.H. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. 2015, 84, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toki, S.; Hara, N.; Ono, K.; Onodera, H.; Tagiri, A.; Oka, S.; Tanaka, H. Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J. 2006, 47, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, T.; Williams, B.A.; Geo, P.; Ali, M.; Gordon, K.; Baren, M.J.; Van Salzberg, S.L.; Wold, B.J.; Lior, P. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar]
- Wang, Y.; Lin, H.; Tong, X.; Hou, Y.; Chang, Y.; Zhang, J. DNA demethylation activates genes in seed maternal integument development in rice (Oryza sativa L.). Plant Physiol. Biochem. 2017, 120, 169–178. [Google Scholar] [CrossRef]
- Ruan, B.; Hua, Z.; Zhao, J.; Zhang, B.; Ren, D.; Liu, C.; Yang, S.; Zhang, A.; Jiang, H.; Yu, H.; et al. OsACL-A2 negatively regulates cell death and disease resistance in rice. Plant Biotechnol. J. 2019, 17, 1344–1356. [Google Scholar] [CrossRef]
- Zhao, J.; Qiu, Z.; Ruan, B.; Kang, S.; He, L.; Zhang, S.; Dong, G.; Hu, J.; Zeng, D.; Zhang, G.; et al. Functional inactivation of putative photosynthetic electron acceptor Ferredoxin C2 (FdC2) induces delayed heading date and decreased photosynthetic rate in rice. PLoS ONE 2015, 10, e0143361. [Google Scholar] [CrossRef]
- Qiu, J.; Hou, Y.; Tong, X.; Wang, Y.; Lin, H.; Liu, Q.; Zhang, W.; Li, Z.; Nallamilli, B.R.; Zhang, J. Quantitative phosphoproteomic analysis of early seed development in rice (Oryza sativa L.). Plant Mol. Biol. 2016, 90, 249–265. [Google Scholar] [CrossRef]
- Kawahara, Y. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Pang, H.; Li, X.; Li, H.; Pan, J.; Chen, W. Long non-coding RNA urothelial cancer-associated 1 promotes bladder cancer cell migration and invasion by way of the hsa-miR-145-ZEB1/2-FSCN1 pathway. Cancer Sci. 2016, 107, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, L.; Yuan, H.; Huang, X.W.; Xiang, T.; Dai, S. Up-regulated lncRNA GAS5 promotes chemosensitivity and apoptosis of triple-negative breast cancer cells. Cell Cycle (Georget. Tex.) 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Golicz, A.A.; Bhalla, P.L.; Singh, M.B. lncRNAs in plant and animal sexual reproduction. Trends Plant Sci. 2018, 23, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ding, Z.; Chen, M.; Yang, G.; Tie, W.; Yan, Y.; Zeng, J.; He, G.; Hu, W. Identification and functional prediction of lncRNAs in response to PEG and ABA treatment in cassava. Environ. Exp. Bot. 2019, 166, 103809. [Google Scholar] [CrossRef]
- Bhatia, G.; Singh, A.; Verma, D.; Sharma, S.; Singh, K. Genome-wide investigation of regulatory roles of lncRNAs in response to heat and drought stress in Brassica juncea (Indian mustard). Environ. Exp. Bot. 2019, 103922. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, W.; Hao, J.; Lv, S.; Wang, C.; Tong, W.; Wang, Y.; Wang, Y.; Liu, X.; Ji, W. Genome-wide identification and functional prediction of novel and fungi-responsive lincRNAs in Triticum aestivum. BMC Genom. 2016, 17, 238. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Shi, S.; Jiang, N.; Khanzada, H.; Wassan, G.M.; Zhu, C.; Peng, X.; Xu, J.; Chen, Y.; Yu, Q.; et al. Genome-wide analysis of long non-coding RNAs affecting roots development at an early stage in the rice response to cadmium stress. BMC Genom. 2018, 19, 460. [Google Scholar] [CrossRef]
- Liu, H.; Wang, R.; Mao, B.; Zhao, B.; Wang, J. Identification of lncRNAs involved in rice ovule development and female gametophyte abortion by genome-wide screening and functional analysis. BMC Genom. 2019, 20, 90. [Google Scholar] [CrossRef]
- Zhao, J.; He, Q.; Chen, G.; Wang, L.; Jin, B. Regulation of non-coding RNAs in heat stress responses of plants. Front. Plant Sci. 2016, 7, 1213. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Cao, R.; Jiao, G.; Lv, Y.; Zhong, M.; Tang, S.; Wei, X.; Hu, P. The involvement of long non-coding RNAs in the formation of high temperature-induced grain chalkiness in rice. Plant Growth Regul. 2018, 86, 263–271. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Sun, F.; Hu, J.; Zha, X.; Su, W.; Yang, J. Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 2018, 9, 3516. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Rao, Y.; Zeng, D.; Yang, Y.; Xu, R.; Zhang, B.; Dong, G.; Qian, Q.; Li, Y. SMALL GRAIN 1, which encodes a mitogen-activated protein kinase kinase 4, influences grain size in rice. Plant J. 2014, 77, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Han, R.; Wu, K.; Zhang, J.; Ye, Y.; Wang, S.; Chen, J.; Pan, Y.; Li, Q.; Xu, X.; et al. G-protein βγ subunits determine grain size through interaction with MADS-domain transcription factors in rice. Nat. Commun. 2018, 9, 852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, P.; Xu, J.; Zeng, D.; Zhang, B.; Geng, M.; Zhang, G.; Huang, K.; Huang, L.; Xu, R.; Ge, S.; et al. Natural variation in the promoter of GSE5 contributes to grain size diversity in rice. Mol. Plant 2017, 10, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Lu, S.J.; Wang, M.J.; He, H.; Sun, L.; Wang, H.; Liu, X.H.; Jiang, L.; Sun, J.L.; Xin, X.; et al. A novel QTL qTGW3 encodes the GSK3/SHAGGY-like kinase OsGSK5/OsSK41 that interacts with OsARF4 to negatively regulate grain size and weight in rice. Mol. Plant 2018, 11, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Qi, P.; Lin, Y.S.; Song, X.J.; Shen, J.B.; Huang, W.; Shan, J.X.; Zhu, M.Z.; Jiang, L.; Gao, J.P.; Lin, H.X. The novel quantitative trait locus GL3.1 controls rice grain size and yield by regulating Cyclin-T1;3. Cell Res. 2012, 22, 1666–1680. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, Y.; Fang, Y.; Zeng, L.; Xu, J.; Yu, H.; Shi, Z.; Pan, J.; Zhang, D.; Kang, S.; et al. A rare allele of GS2 enhances grain size and grain yield in rice. Mol. Plant 2015, 8, 1455–1465. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Sun, S.; Yao, J.; Wang, C.; Yu, S.; Xu, C.; Li, X.; Zhang, Q. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc. Natl. Acad. Sci. USA 2010, 107, 19579–19584. [Google Scholar] [CrossRef] [Green Version]
- Song, X.J.; Huang, W.; Shi, M.; Zhu, M.Z.; Lin, H.X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 2007, 39, 623–630. [Google Scholar] [CrossRef]
- Liu, J.; Chen, J.; Zheng, X.; Wu, F.; Lin, Q.; Heng, Y.; Tian, P.; Cheng, Z.; Yu, X.; Zhou, K.; et al. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 2017, 3, 17043. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEG Set | DELncs Number | Up-Regulated | Down-Regulated |
|---|---|---|---|
| 0 DAP vs 3 DAP | 447 | 190 | 257 |
| 0 DAP vs 7 DAP | 454 | 218 | 236 |
| 3 DAP vs 7 DAP | 440 | 246 | 194 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Ajadi, A.A.; Wang, Y.; Tong, X.; Wang, H.; Tang, L.; Li, Z.; Shu, Y.; Liu, X.; Li, S.; et al. Genome-Wide Identification of lncRNAs During Rice Seed Development. Genes 2020, 11, 243. https://doi.org/10.3390/genes11030243
Zhao J, Ajadi AA, Wang Y, Tong X, Wang H, Tang L, Li Z, Shu Y, Liu X, Li S, et al. Genome-Wide Identification of lncRNAs During Rice Seed Development. Genes. 2020; 11(3):243. https://doi.org/10.3390/genes11030243
Chicago/Turabian StyleZhao, Juan, Abolore Adijat Ajadi, Yifeng Wang, Xiaohong Tong, Huimei Wang, Liqun Tang, Zhiyong Li, Yazhou Shu, Xixi Liu, Shufan Li, and et al. 2020. "Genome-Wide Identification of lncRNAs During Rice Seed Development" Genes 11, no. 3: 243. https://doi.org/10.3390/genes11030243