Geographic Life History Differences Predict Genomic Divergence Better than Mitochondrial Barcodes or Phenotype

Abstract
:1. Introduction
2. Materials and Methods
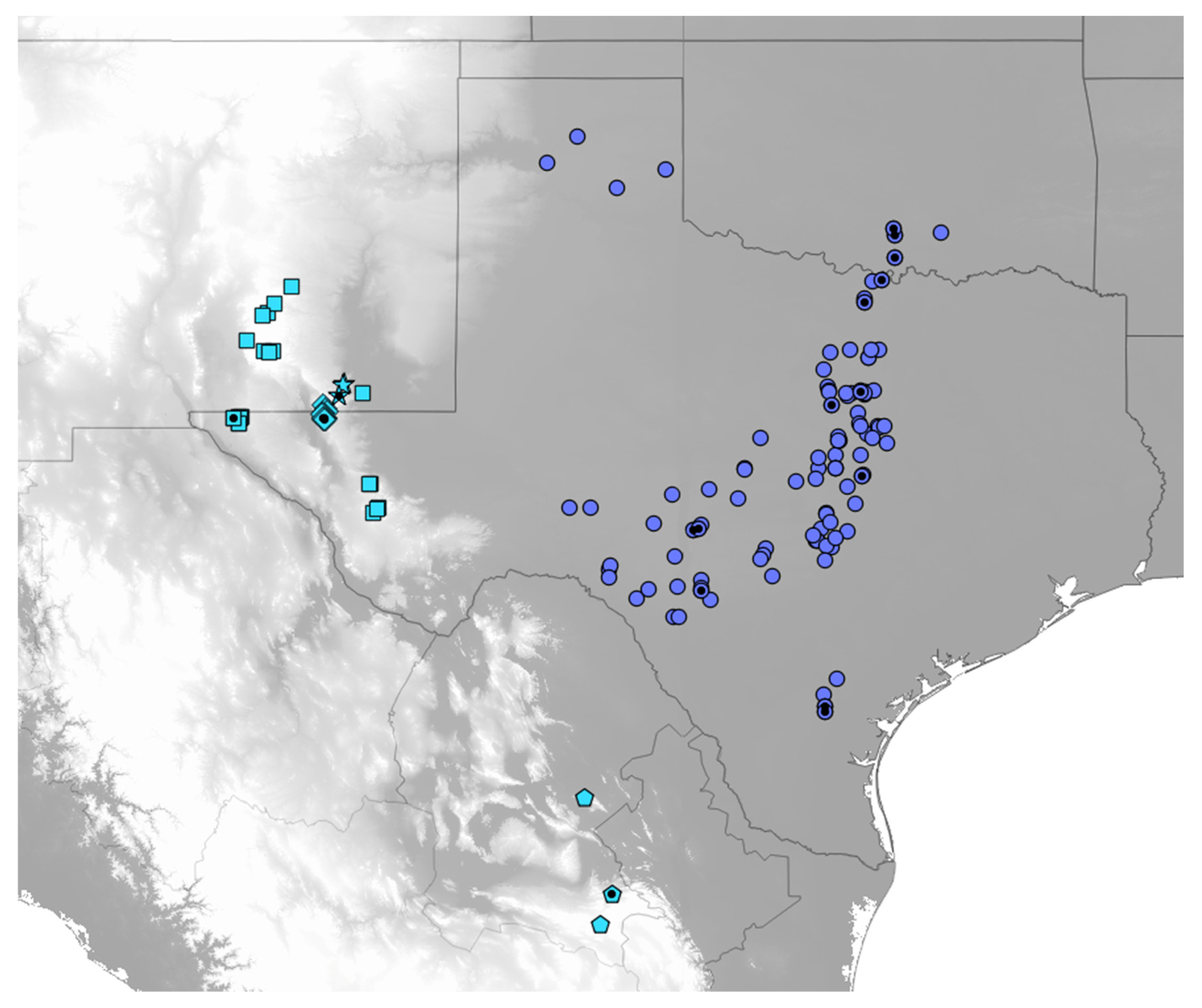
2.1. Specimen Collection and Distribution Data
2.2. Molecular Sampling, mtDNA
2.3. Mitochondrial Genealogy
2.4. Multilocus Marker Generation and Analysis
2.5. Principal Component Analysis of SNP Data
2.6. Bayesian Clustering Analysis Using SNP Data
2.7. Multi-Locus Nuclear Trees Generated from SNP Data
2.8. Phenology Analyses
3. Results
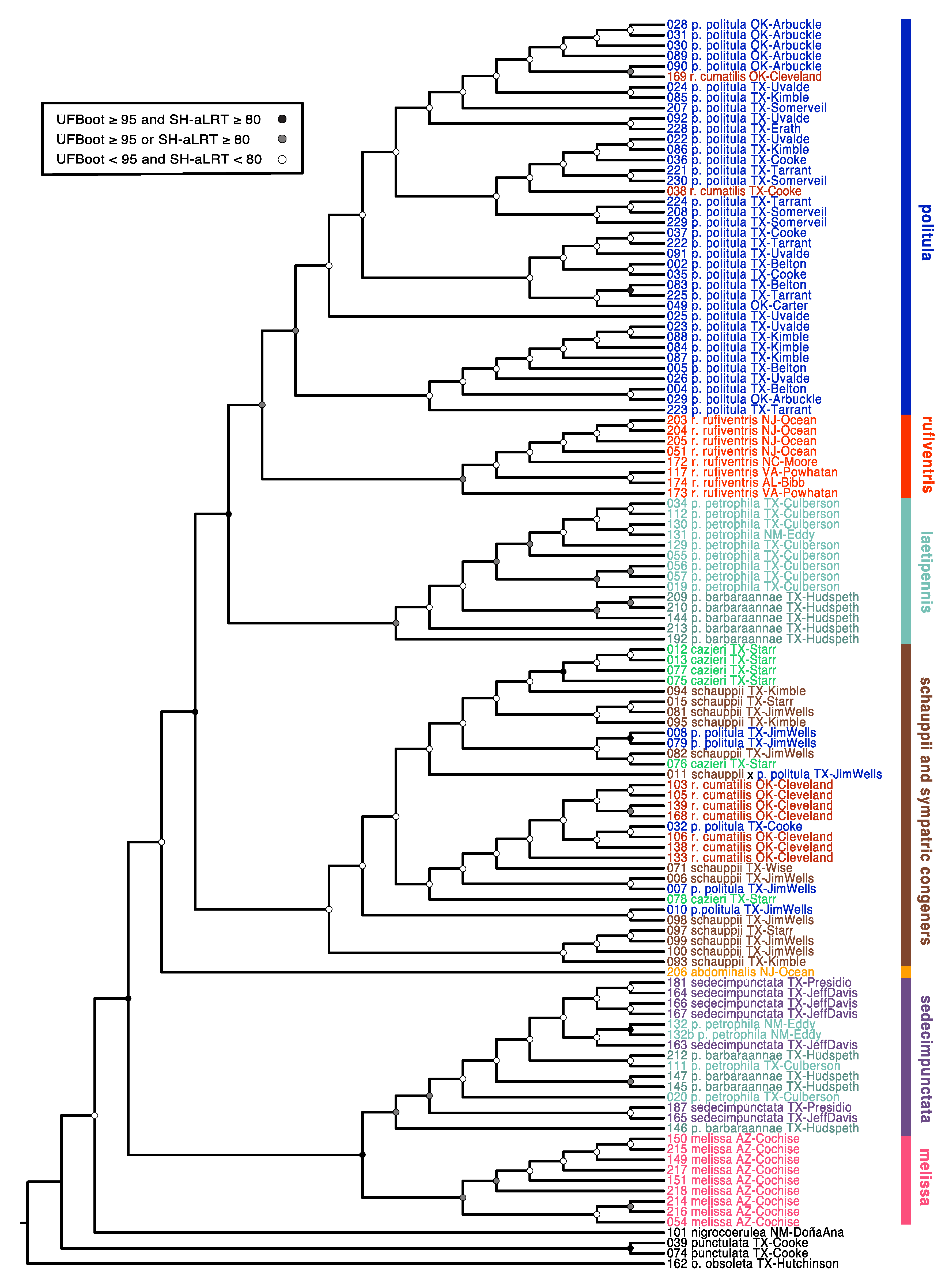
3.1. Mitochondrial Genealogies
3.2. Maximum Likelihood Topologies from SNP Data
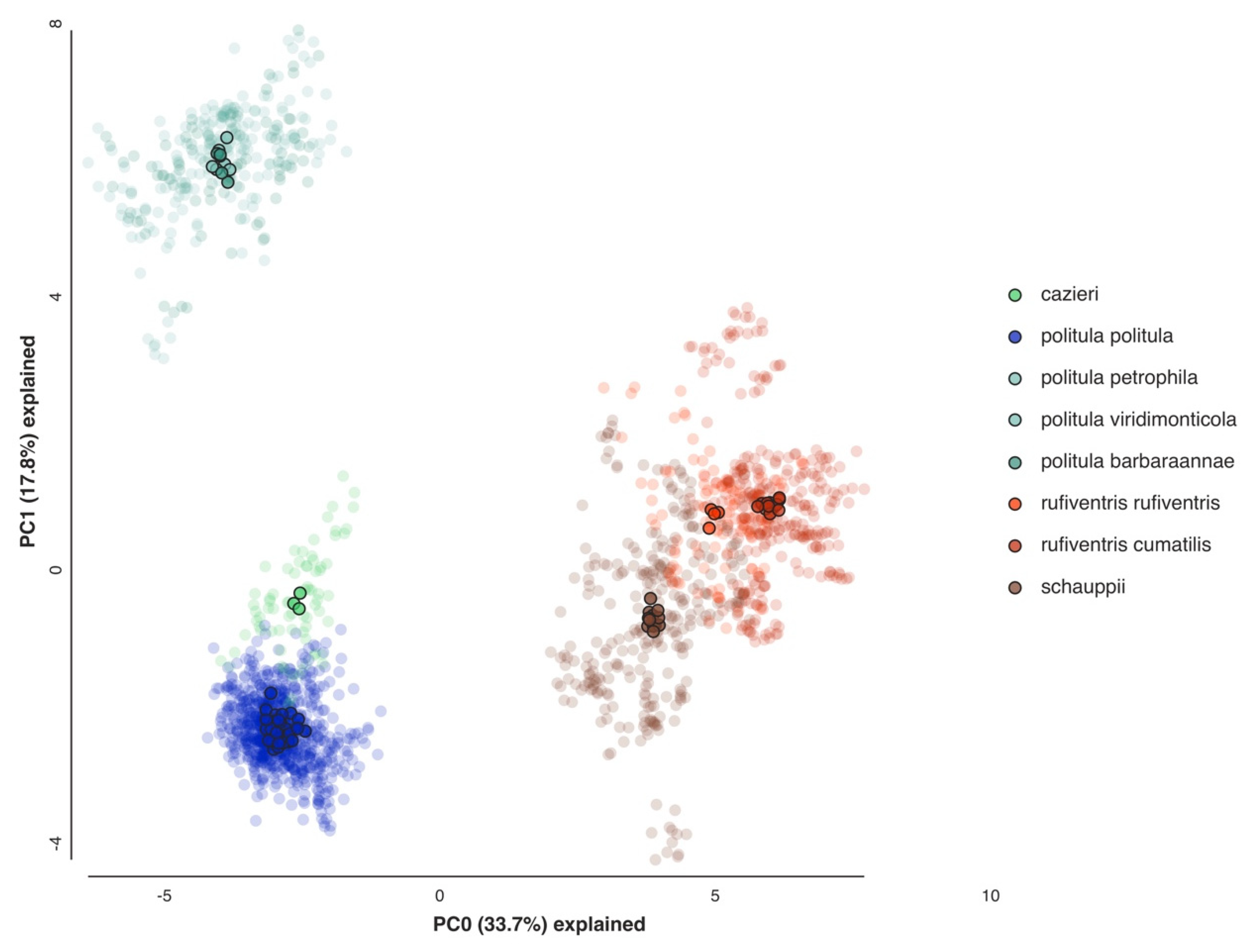
3.3. PCA of Multilocus Data
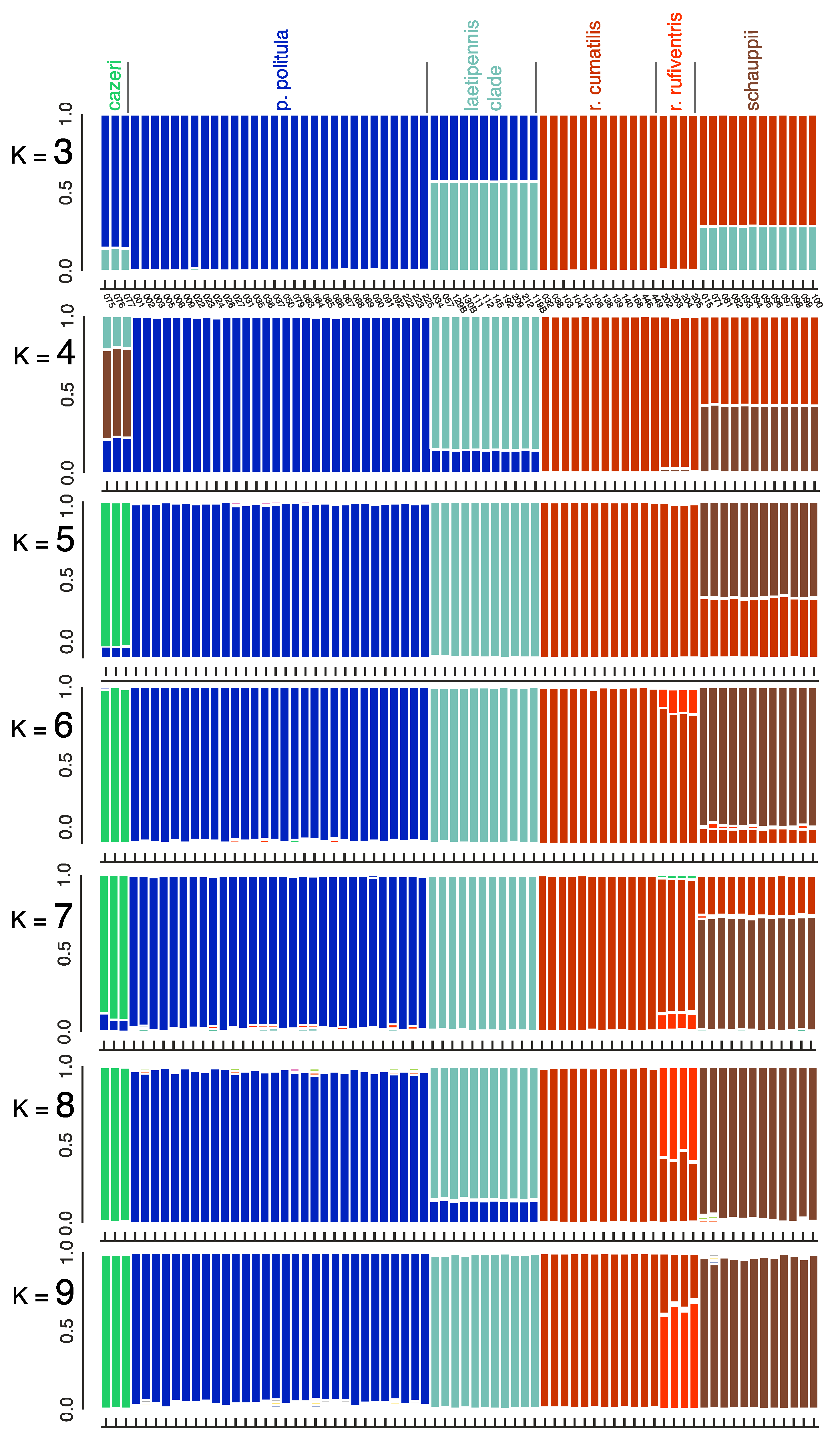
3.4. Bayesian Clustering Analysis
3.5. Phenology and Geography
3.6. Taxonomy
4. Discussion
4.1. Incongruence of Phenotype and Taxonomy with Multilocus Results
4.2. Phenotypic Plasticity of Color
4.3. Mito-Nuclear Discordance, and Interpretation of mtDNA Genealogies for Species Identification
4.4. Hybridization, Introgression, and ‘Mitochondrial Displacement’
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johannsen, W. The genotype conception of heredity. Am. Nat. 1911, 45, 129–159. [Google Scholar] [CrossRef] [Green Version]
- Linne, C.R.; Poh, Y.-P.; Peterson, B.K.; Barrett, R.D.H.; Larson, J.G.; Jensen, J.D.; Hoekstra, H.E. Adaptive evolution of multiple traits through multiple mutations at a single gene. Science 2013, 339, 1312–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, R.D.H.; Laurent, S.; Mallarino, R.; Pfeifer, S.P.; Xu, C.C.Y.; Foll, M.; Wakamatsu, K.; Duke-Cohan, J.S.; Jensen, J.D.; Hoekstra, H.E. Linking a mutation to survival in wild mice. Science 2019, 363, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Knisley, C.B.; Schultz, T.D. The Biology of Tiger Beetles and a Guide to the Species of the South Atlantic States; Virginia Museum of Natural History Special Collection Number 5: Martinsville, VA, USA, 1997; pp. 1–210. [Google Scholar]
- Pearson, D.L.; Cassola, F. A quantitative analysis of species descriptions of tiger beetles (Coleoptera: Cicindelinae), from 1758 to 2004, and notes about related developments in biodiversity studies. Coleop. Bull. 2005, 59, 184–193. [Google Scholar] [CrossRef]
- Duran, D.P.; Herrmann, D.P.; Roman, S.J.; Gwiazdowski, R.A.; Drummond, J.A.; Hood, G.R.; Egan, S.P. Cryptic diversity in the North American Dromochorus tiger beetles (Coleoptera: Carabidae: Cicindelinae): A congruence-based method for species discovery. Zool. J. Linn. Soc.-Lond. 2019, 186, 250–285. [Google Scholar] [CrossRef]
- Willis, H.L. Artificial key to the species of Cicindela of North America north of Mexico (Coleoptera: Cicindelidae). J. Kans. Entomol. Soc. 1968, 41, 303–317. [Google Scholar]
- Pearson, D.L.; Knisley, C.B.; Duran, D.P.; Kazilek, C.J. A Field Guide to Tiger Beetles of the United States and Canada: Identification, Natural History, and Distribution of the Cicindelidae; Oxford University Press: New York, NY, USA, 2015; pp. 1–251. [Google Scholar]
- Knisley, C.B.; Kippenhan, M.; Brzoska, D. Conservation status of United States tiger beetles. Terr. Arthropod Rev. 2014, 7, 93–145. [Google Scholar] [CrossRef] [Green Version]
- Shelford, V.E. Color and color-pattern mechanism of tiger beetles. Ill. Biol. Monogr. 1917, 3, 395–532. [Google Scholar]
- Shaw, K.L.; Mullen, S.P. Speciation continuum. J. Hered. 2014, 105, 741–742. [Google Scholar] [CrossRef]
- Mayr, E. Systematics and the Origin of Species; Columbia University Press: New York, NY, USA, 1942. [Google Scholar]
- Mayr, E. Animal Species and Evolution; Harvard University Press: Cambridge, MA, USA, 1963. [Google Scholar]
- Mallet, J. Subspecies, semispecies, superspecies. In Encyclopedia of Biodiversity; Levin, S., Ed.; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Schultz, T.D. The ultrastructure, physiology, and ecology of epicuticular interference reflectors in tiger beetles (Cicindela). Ph.D. Thesis, The University of Texas at Austin, Austin, TX, USA, 1983. [Google Scholar]
- Wiesner, J. Verzeichnis der Sandlaufkäfer der Welt (Coleoptera, Cicindelidae); Verlag Erna Bauer: Keltern, Germany, 1992; Volume 27, pp. 1–364. [Google Scholar]
- Freitag, R. Catalogue of the Tiger Beetles of Canada and the United States; NRC Research Press: Ottawa, ON, Canada, 1999; pp. 1–195. [Google Scholar]
- Pearson, D.L.; Knisley, C.B.; Kazilek, C.J. A Field Guide to Tiger Beetles of the United States and Canada: Identification, Natural History, and Distribution of the Cicindelidae; Oxford University Press: New York, NY, USA, 2006; pp. 1–27. [Google Scholar]
- Bousquet, Y. Catalogue of Geadephaga (Coleoptera, Adephaga) of America, north of Mexico. ZooKeys 2012, 245, 1–1630. [Google Scholar] [CrossRef] [Green Version]
- Horn, W. 50 neue Cicindelinae. Arch. FüR Nat. 1913, A11, 1–33. [Google Scholar]
- Sumlin, W.D. A new Subspecies of Cicindela politula from west Texas and a note on C. cazieri (Coleoptera; Cicindelidae). J. Kans. Entomol. Soc. 1976, 49, 521–526. [Google Scholar]
- Sumlin, W.D. A review of Cicindela politula LeConte (Coleoptera: Cicindelidae). J. Kans. Entomol. Soc. 1985, 58, 220–227. [Google Scholar]
- Gage, E.V. A new subspecies of Cicindela politula from New Mexico and a range extension for Cicindela politula barbaraannae (Coleoptera: Cicindelidae). Entomol. News 1988, 99, 143–147. [Google Scholar]
- Murray, R.R.; Acciavatti, R.E. Notes on the habitat, distribution and timing of occurrence of Cicindela politula LeConte. Cicindela 1976, 8, 1–5. [Google Scholar]
- Knowles, L.L. Tests of Pleistocene Speciation in montane grasshoppers (genus Melanoplus) from the Sky Islands of Western North America. Evolution 2000, 54, 1337–1348. [Google Scholar] [CrossRef]
- Cazier, M.A. Two new western tiger beetles, with notes (Coleoptera-Cicindelidae). Bull. Brooklyn Entomol. Soc. 1939, 34, 24–28. [Google Scholar]
- Cazier, M.A. A review of the Mexican tiger beetles of the genus Cicindela (Coleoptera, Cicindelidae). B Am. Mus. Nat. Hist. 1954, 103, 231–309. [Google Scholar]
- Sumlin, W.D.; San Antonio, TX, USA. Personal Communication, 2019.
- Funk, D.J.; Omland, K.E. Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Ann. Rev. Ecol. Evol. S 2003, 34, 397–423. [Google Scholar] [CrossRef] [Green Version]
- Duran, D.P. Speciation and diversification in the North American tiger beetles of the Cicindela sylvatica group: Morphological variation and an ecophylogeographic approach. Ph.D. Thesis, Vanderbilt University, Nashville, TN, USA, 2010. [Google Scholar]
- Vogler, A.P.; Cardoso, A.; Barraclough, T. Exploring rate variation among and within sites in a densely sampled tree: Species-level phylogenetics of North American tiger beetles (genus Cicindela). Syst. Biol. 2005, 54, 4–20. [Google Scholar] [CrossRef] [Green Version]
- Gough, H.M.; Duran, D.P.; Kawahara, A.Y.; Toussaint, E.F.A. A comprehensive molecular phylogeny of tiger beetles (Coleoptera, Carabidae, Cicindelinae). Syst. Entomol. 2019, 44, 305–321. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Hebert, P.D.; Ratnasingham, S.; De Waard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. Lond. B-Biol. Sci. 2003, 270S, S96–S99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crozier, R.H.; Crozier, Y.C. The cytochrome b and ATPase genes of honeybee mitochondrial DNA. Mol. Biol. Evol. 1992, 9, 474–482. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Gompert, Z.; Mudge, J.; Schilkey, F.D.; Benkman, C.W.; Buerkle, C.A. Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol. Ecol. 2012, 21, 2991–3005. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010, 6, e1000862. [Google Scholar] [CrossRef] [Green Version]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.A.R. PyRAD: Assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 2014, 30, 1844–1849. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Dray, S.; Josse, J. Principal component analysis with missing values: A comparative survey of methods. Plant Ecol. 2015, 216, 657–667. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, K.J.; Andrew, R.L.; Bock, D.G.; Franklin, M.T.; Kane, N.C.; Moore, J.-S.; Moyers, B.T.; Renaut, S.; Rennison, D.J.; Veen, T.; et al. Recommendations for utilizing and reporting population genetic analyses: The reproducibility of genetic clustering using the progrAm. structure. Mol. Ecol. 2012, 21, 4925–4930. [Google Scholar] [CrossRef]
- Janes, J.K.; Miller, J.M.; Dupuis, J.R.; Malefant, R.M.; Gorrell, J.C.; Cullingham, C.I.; Andrew, R.L. The K = 2 conundrum. Mol. Ecol. 2017, 26, 3594–3602. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2007, 22, 148–155. [Google Scholar] [CrossRef]
- Gwiazdowski, R.A.; Vea, I.M.; Andersen, J.C.; Normark, B.B. Discovery of cryptic species among North American pine-feeding Chionaspis scale insects (Hemiptera: Diaspididae). Biol. J. Linn. Soc. 2011, 104, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Duran, D.P.; Gough, H.M. Unifying systematics and taxonomy: Nomenclatural changes to Nearctic tiger beetles (Coleoptera: Carabidae: Cicindelinae) based on phylogenetics, morphology and life history. Insect Mun. 2019, 0727, 1–12. [Google Scholar]
- Rivalier, E. Démembrement du genre Cicindela Linné. II. Faune américaine. Rev. Fr. Entomol. 1954, 21, 249–268. [Google Scholar]
- Boyd, H.P. Checklist of Cicindelidae: The Tiger Beetles; Marlton, N.J. Plexus Publ., Inc.: Marlton, NJ, USA, 1982; pp. 1–31. [Google Scholar]
- Vogt, G. Three new Cicindelidae from south Texas with collecting notes on other Cicindelidae (Coleoptera). Bull. Brooklyn Entomol. Soc. 1949, 44, 1–9. [Google Scholar]
- Schultz, T.D.; Rankin, M.A. The ultrastructure of the epicuticular interference reflectors of tiger beetles (Cicindela). J. Exp. Biol. 1985, 117, 87–110. [Google Scholar]
- Schultz, T.D.; Rankin, M.A. Development changes in the interference reflectors and coloration of tiger beetles (Cicindela). J. Exp. Biol. 1985, 117, 111–117. [Google Scholar]
- Schultz, T.D.; Bernard, G.D. Pointillistic mixing of interference colours in cryptic tiger beetles. Nature 1989, 337, 72–73. [Google Scholar] [CrossRef]
- Willis, H.L. Bionomics and zoogeography of tiger beetles of saline habitats in the central United States (Coleoptera: Cicindelidae). Univ. Kans. Sci. Bull. 1967, 47, 145–313. [Google Scholar]
- Schultz, T.D. Role of structural colors in predator avoidance by tiger beetles of the genus Cicindela (Coleoptera: Cicindelidae). Bull. Entomol. Soc. Am. 1986, 32, 142–146. [Google Scholar] [CrossRef]
- Acorn, J.A. Mimetic tiger beetles and the puzzle of cicindelid coloration. Coleop. Bull. 1988, 42, 28–33. [Google Scholar]
- Pearson, D.L. Biology of tiger beetles. Ann. Rev. Entomol. 1988, 33, 123–147. [Google Scholar] [CrossRef]
- Schultz, T.D.; Hadley, N.F. Structural colors of tiger beetles and their role in heat transfer through the integument. Physiol. Zool. 1987, 60, 737–745. [Google Scholar] [CrossRef]
- Acorn, J.A. The historical development of geographic color variation among dune Cicindela in western Canada. In The Biogeography of Ground Beetles of Mountains and Islands; Noonan, G.E., Ball, G.E., Stork, N.E., Eds.; Intercept Press: Andover, UK, 1992; pp. 217–233. [Google Scholar]
- Schultz, T.D.; Quinlan, M.C.; Hadley, N.F. Preferred body temperature, metabolic physiology, and water balance of adult Cicindela longilabris: A comparison of populations from boreal habitats and climatic refugia. Physiol. Zool. 1992, 65, 226–242. [Google Scholar] [CrossRef]
- Sperling, F.A.H. DNA barcoding: Deus ex machina. Newsl. Biol. Surv. Can. 2003, 22, 1–3. [Google Scholar]
- Gompert, Z.; Nice, C.C.; Fordyce, J.A.; Forister, M.L.; Shapiro, A.M. Identifying units for conservation using molecular systematics: The cautionary tale of the Karner blue butterfly. Mol. Ecol. 2006, 15, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Rubinoff, D.; Cameron, S.; Will, K. A genomic perspective on the shortcomings of mitochondrial DNA for “barcoding” identification. J. Hered. 2006, 97, 581–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan-Richards, M.; Bulgarella, M.; Sivyer, L.; Dowle, E.J.; Hale, M.; McKean, N.E.; Trewick, S.A. Explaining large mitochondrial sequence differences within a population sample. R. Soc. Open Sci. 2017, 4, 170730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.; Hubricht, L. Hybridization in Tradescantia. III. The evidence for introgressive hybridization. Am. J. Bot. 1938, 25, 396–402. [Google Scholar] [CrossRef]
- Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 2005, 20, 229–237. [Google Scholar] [CrossRef]
- Fitzpatrick, B.M.; Johnson, J.R.; Kump, D.K.; Smith, J.J.; Voss, S.R.; Shaffer, H.B. Rapid spread of invasive genes into a threatened native species. Proc. Nat. Acad. Sci. USA 2010, 107, 3606–3610. [Google Scholar] [CrossRef] [Green Version]
- Jiggins, F.M. Male-killing Wolbachia and mitochondrial DNA: Selective sweeps, hybrid introgression and parasite population dynamics. Genetics 2003, 164, 5–12. [Google Scholar]
- Jones, T.; Conner, W.E. Pre-mating reproductive isolation in tiger beetles (Carabidae: Cicindelinae): An examination of the role of visual and morphological feedback. J. Insect Behav. 2018, 31, 672–688. [Google Scholar] [CrossRef]
- Freitag, R. Selection for a non-genitalic mating structure in female tiger beetles of the genus Cicindela (Coleoptera: Cicindelidae). Can. Entomol. 1974, 106, 561–568. [Google Scholar] [CrossRef]
- Eberhard, W.G. Sexual Selection and Animal Genitalia; Harvard University Press: Cambridge, MA, USA, 1985. [Google Scholar]
- Freitag, R. Revision of the North American species of the Cicindela maritima group with a study of hybridization between Cicindela duodecimguttata and oregona. Quaest. Entomol. 1965, 1, 87–170. [Google Scholar]
- Gwiazdowski, R.A.; Gillespie, S.; Weddle, R.; Elkinton, J. Laboratory rearing of common and endangered species of North American tiger beetles (Coleoptera: Carabidae: Cicindelinae). Ann. Entomol. Soc. Am. 2011, 104, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Schincariol, L.A. Mating behavior, spermatophore structure, ecology and systematics of the Cicindela splendida group (Coleoptera: Cicindelidae). Master Thesis, Lakehead University, Thunder Bay, ON, Canada, 1988; pp. 1–236. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Estimate | Std Error | t Ratio | Prob > |t| |
|---|---|---|---|---|
| Intercept | 243.86075 | 21.31486 | 11.44 | <0.0001 |
| Clade | −29.31527 | 1.239944 | −23.64 | <0.0001 |
| Geography | 0.8245943 | 0.678734 | 1.21 | 0.2259 |
| Clade × Geography | 2.3503926 | 0.678734 | 3.46 | 0.0007 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duran, D.P.; Laroche, R.A.; Gough, H.M.; Gwiazdowski, R.A.; Knisley, C.B.; Herrmann, D.P.; Roman, S.J.; Egan, S.P. Geographic Life History Differences Predict Genomic Divergence Better than Mitochondrial Barcodes or Phenotype. Genes 2020, 11, 265. https://doi.org/10.3390/genes11030265
Duran DP, Laroche RA, Gough HM, Gwiazdowski RA, Knisley CB, Herrmann DP, Roman SJ, Egan SP. Geographic Life History Differences Predict Genomic Divergence Better than Mitochondrial Barcodes or Phenotype. Genes. 2020; 11(3):265. https://doi.org/10.3390/genes11030265
Chicago/Turabian StyleDuran, Daniel P., Robert A. Laroche, Harlan M. Gough, Rodger A. Gwiazdowski, Charles B. Knisley, David P. Herrmann, Stephen J. Roman, and Scott P. Egan. 2020. "Geographic Life History Differences Predict Genomic Divergence Better than Mitochondrial Barcodes or Phenotype" Genes 11, no. 3: 265. https://doi.org/10.3390/genes11030265
APA StyleDuran, D. P., Laroche, R. A., Gough, H. M., Gwiazdowski, R. A., Knisley, C. B., Herrmann, D. P., Roman, S. J., & Egan, S. P. (2020). Geographic Life History Differences Predict Genomic Divergence Better than Mitochondrial Barcodes or Phenotype. Genes, 11(3), 265. https://doi.org/10.3390/genes11030265