Dissecting the Genetic Basis of Variation in Drosophila Sleep Using a Multiparental QTL Mapping Resource

Abstract
:1. Introduction
2. Materials and Methods
2.1. Mapping Population
2.2. Phenotyping Assay
2.3. QTL Mapping in the DSPR
2.4. Correlations Among Strain Effects at QTL
2.5. Functional Testing via RNAi
2.6. Data and Scripts
3. Results and Discussion
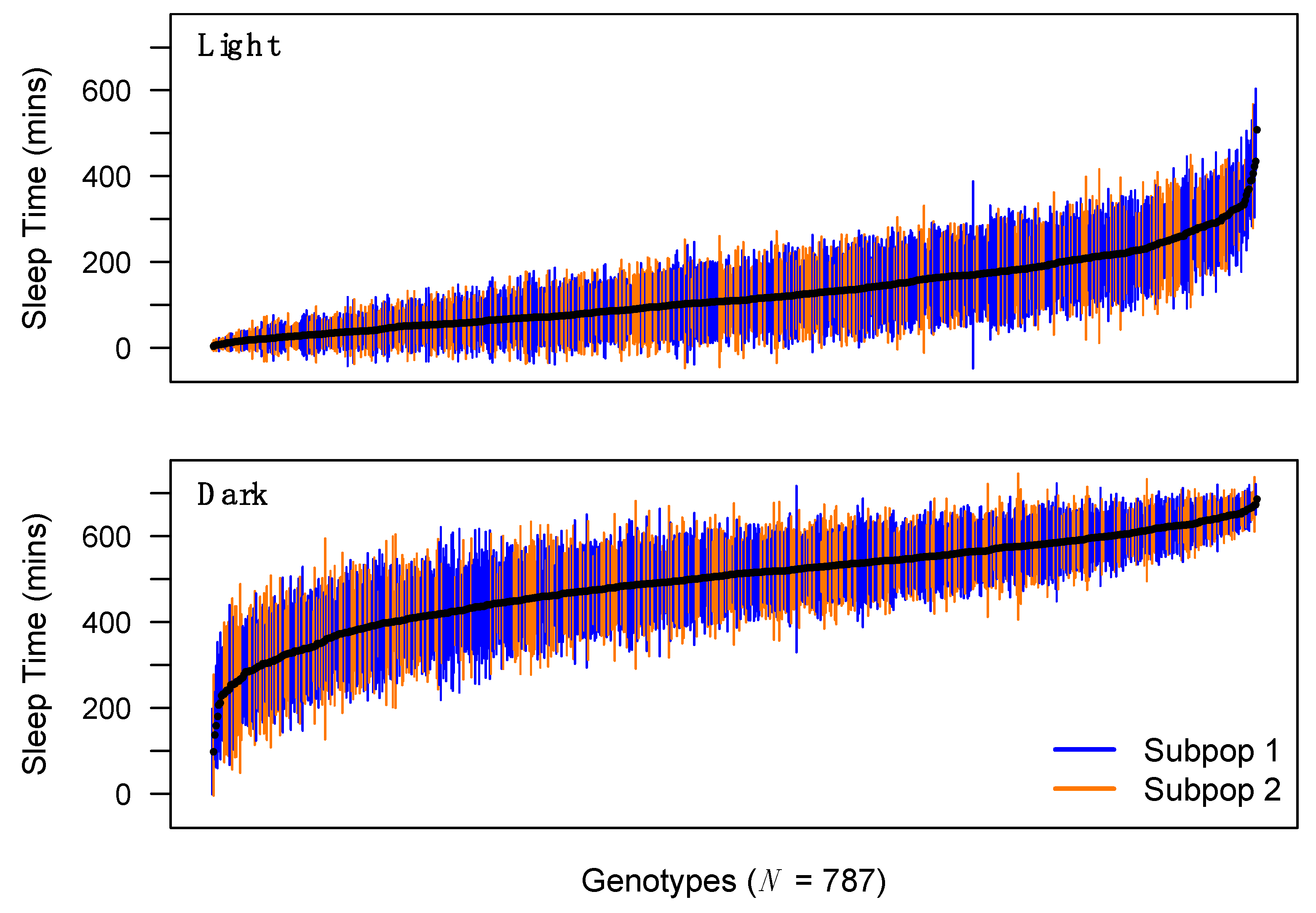
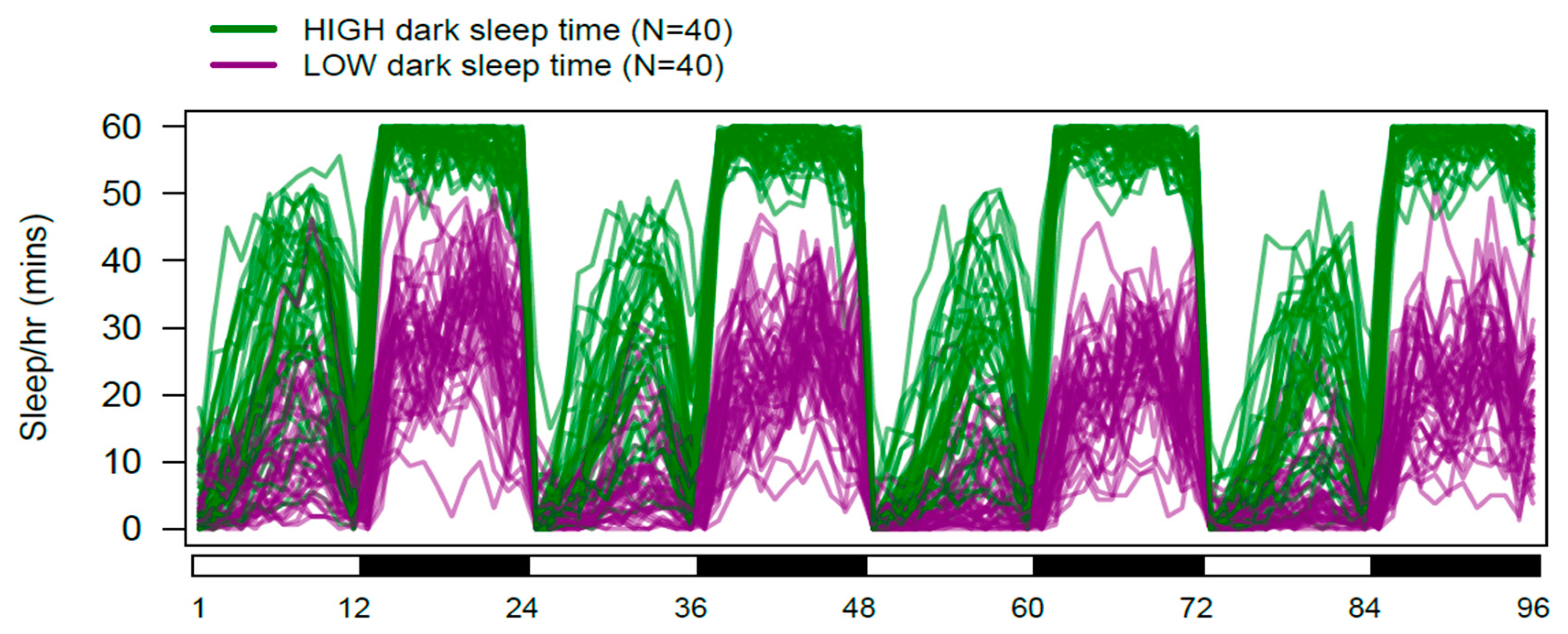
3.1. Extensive Genetic Variation for Sleep and Activity Traits in the DSPR
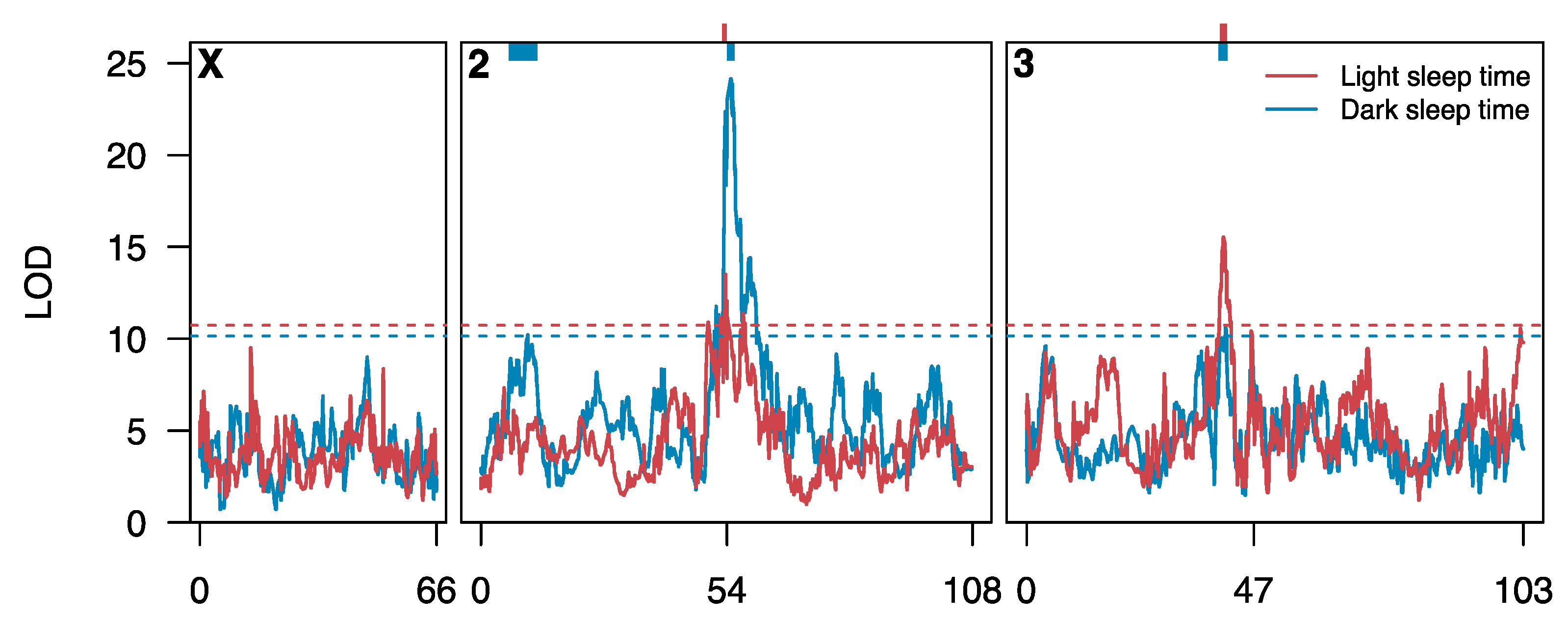
3.2. Loci Contributing to Genetic Variation in Sleep
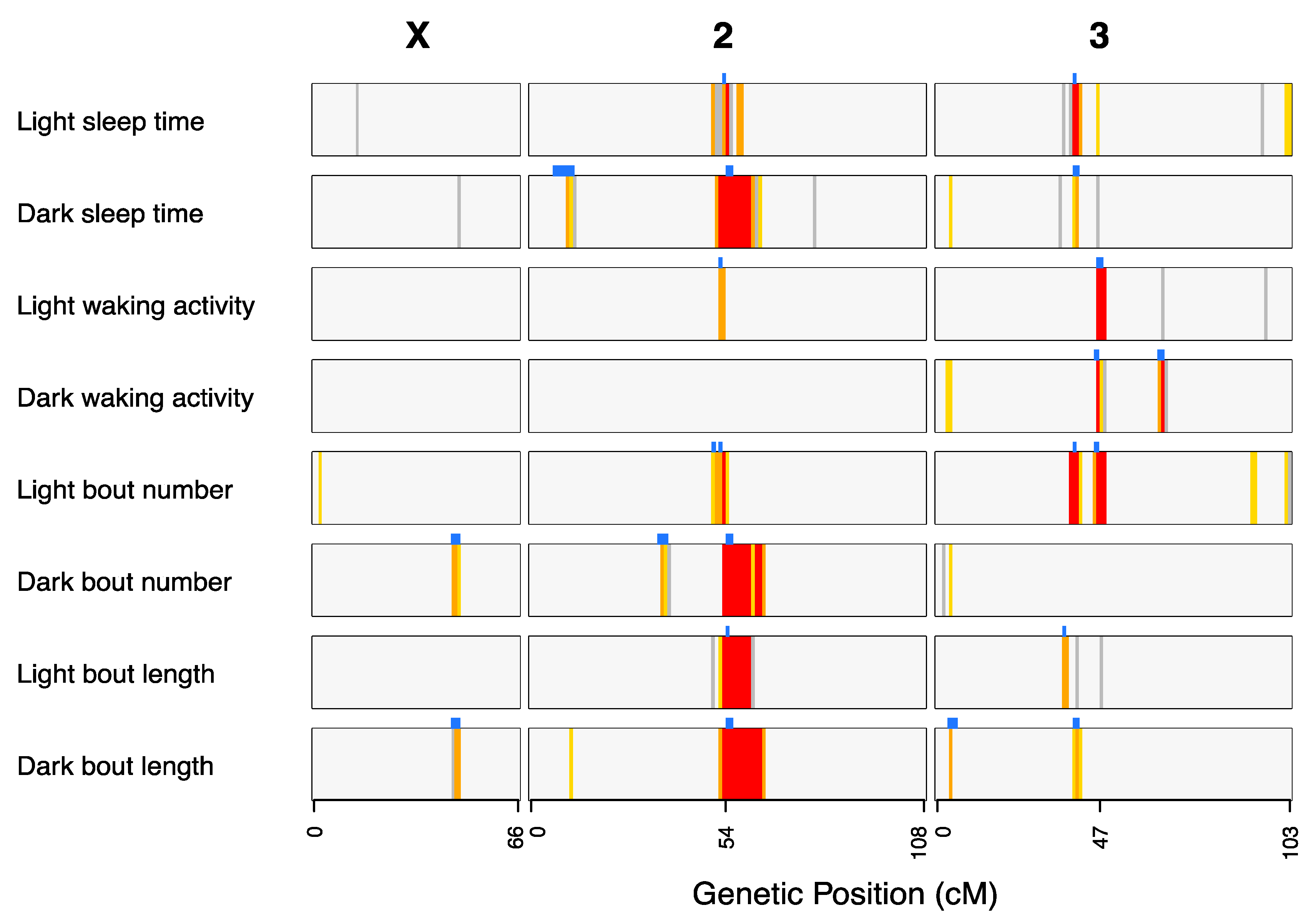
3.3. QTL Overlap Among Traits
3.4. Resolving Plausible Regulatory Genes via eQTL Mapping
3.5. Correlations between Gene Expression and Phenotype
3.6. Overlap with Loci Previously Implicated in the Genetic Control of Natural Variation in Sleep in Flies
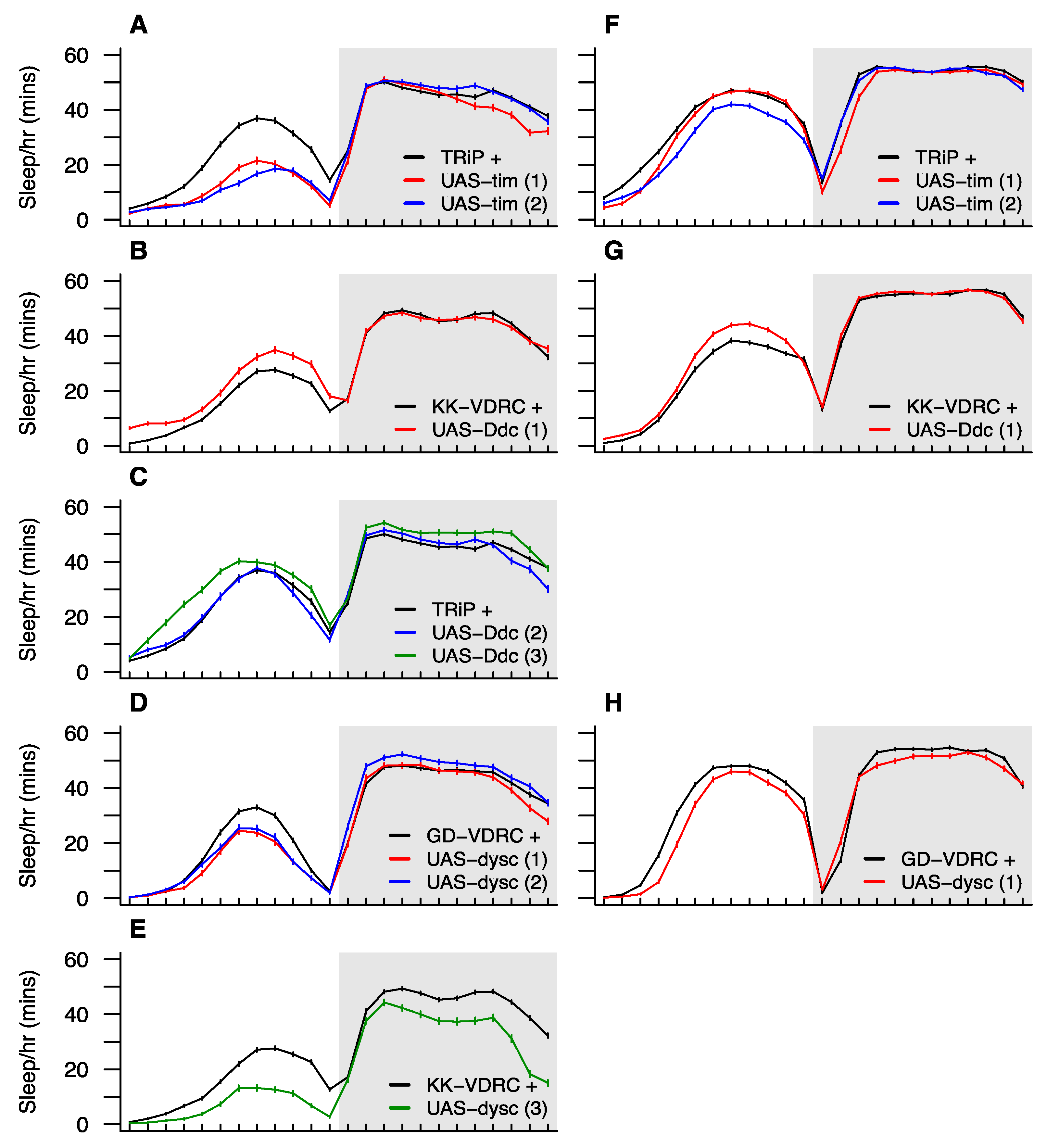
3.7. Functional Testing of Candidate Sleep Genes via Neuron-Specific RNAi
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allada, R.; Siegel, J.M. Unearthing the phylogenetic roots of sleep. Curr. Biol. 2008, 18, R670–R679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.S.; Tobler, I. Animal sleep: A review of sleep duration across phylogeny. Neurosci. Biobehav. Rev. 1984, 8, 269–300. [Google Scholar] [CrossRef]
- Cirelli, C.; Tononi, G. Is sleep essential? PLoS Biol. 2008, 6, e216. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.M. Do all animals sleep? Trends Neurosci. 2008, 31, 208–213. [Google Scholar] [CrossRef]
- Winrow, C.J.; Williams, D.L.; Kasarskis, A.; Millstein, J.; Laposky, A.D.; Yang, H.S.; Mrazek, K.; Zhou, L.; Owens, J.R.; Radzicki, D.; et al. Uncovering the genetic landscape for multiple sleep-wake traits. PLoS ONE 2009, 4, e5161. [Google Scholar] [CrossRef]
- Harbison, S.T.; McCoy, L.J.; Mackay, T.F. Genome-wide association study of sleep in Drosophila melanogaster. BMC Genomics 2013, 14, 281. [Google Scholar] [CrossRef] [Green Version]
- Svetec, N.; Zhao, L.; Saelao, P.; Chiu, J.C.; Begun, D.J. Evidence that natural selection maintains genetic variation for sleep in Drosophila melanogaster. BMC Evol. Biol. 2015, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Dashti, H.S.; Jones, S.E.; Wood, A.R.; Lane, J.M.; van Hees, V.T.; Wang, H.; Rhodes, J.A.; Song, Y.; Patel, K.; Anderson, S.G.; et al. Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nat. Commun. 2019, 10, 1100. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Shmygelska, A.; Tran, D.; Eriksson, N.; Tung, J.Y.; Hinds, D.A. GWAS of 89,283 individuals identifies genetic variants associated with self-reporting of being a morning person. Nat. Commun. 2016, 7, 10448. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.E.; Tyrrell, J.; Wood, A.R.; Beaumont, R.N.; Ruth, K.S.; Tuke, M.A.; Yaghootkar, H.; Hu, Y.; Teder-Laving, M.; Hayward, C.; et al. Genome-Wide Association Analyses in 128,266 Individuals Identifies New Morningness and Sleep Duration Loci. PLoS Genet. 2016, 12, e1006125. [Google Scholar] [CrossRef]
- Wang, H.; Lane, J.M.; Jones, S.E.; Dashti, H.S.; Ollila, H.M.; Wood, A.R.; van Hees, V.T.; Brumpton, B.; Winsvold, B.S.; Kantojarvi, K.; et al. Genome-wide association analysis of self-reported daytime sleepiness identifies 42 loci that suggest biological subtypes. Nat. Commun. 2019, 10, 3503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.E.; Lane, J.M.; Wood, A.R.; van Hees, V.T.; Tyrrell, J.; Beaumont, R.N.; Jeffries, A.R.; Dashti, H.S.; Hillsdon, M.; Ruth, K.S.; et al. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat. Commun. 2019, 10, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendricks, J.C.; Finn, S.M.; Panckeri, K.A.; Chavkin, J.; Williams, J.A.; Sehgal, A.; Pack, A.I. Rest in Drosophila is a sleep-like state. Neuron 2000, 25, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.J.; Cirelli, C.; Greenspan, R.J.; Tononi, G. Correlates of sleep and waking in Drosophila melanogaster. Science 2000, 287, 1834–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubowy, C.; Sehgal, A. Circadian Rhythms and Sleep in Drosophila melanogaster. Genetics 2017, 205, 1373–1397. [Google Scholar] [CrossRef] [Green Version]
- Cirelli, C.; Bushey, D.; Hill, S.; Huber, R.; Kreber, R.; Ganetzky, B.; Tononi, G. Reduced sleep in Drosophila Shaker mutants. Nature 2005, 434, 1087–1092. [Google Scholar] [CrossRef]
- Farca Luna, A.J.; Perier, M.; Seugnet, L. Amyloid Precursor Protein in Drosophila Glia Regulates Sleep and Genes Involved in Glutamate Recycling. J. Neurosci. 2017, 37, 4289–4300. [Google Scholar] [CrossRef] [Green Version]
- Koh, K.; Joiner, W.J.; Wu, M.N.; Yue, Z.; Smith, C.J.; Sehgal, A. Identification of SLEEPLESS, a sleep-promoting factor. Science 2008, 321, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffenberger, C.; Allada, R. Cul3 and the BTB adaptor insomniac are key regulators of sleep homeostasis and a dopamine arousal pathway in Drosophila. PLoS Genet. 2012, 8, e1003003. [Google Scholar] [CrossRef] [Green Version]
- Toda, H.; Williams, J.A.; Gulledge, M.; Sehgal, A. A sleep-inducing gene, nemuri, links sleep and immune function in Drosophila. Science 2019, 363, 509–515. [Google Scholar] [CrossRef]
- Harbison, S.T.; Carbone, M.A.; Ayroles, J.F.; Stone, E.A.; Lyman, R.F.; Mackay, T.F. Co-regulated transcriptional networks contribute to natural genetic variation in Drosophila sleep. Nat. Genet. 2009, 41, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbison, S.T.; Serrano Negron, Y.L.; Hansen, N.F.; Lobell, A.S. Selection for long and short sleep duration in Drosophila melanogaster reveals the complex genetic network underlying natural variation in sleep. PLoS Genet. 2017, 13, e1007098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, E.G.; Macdonald, S.J.; Long, A.D. Properties and power of the Drosophila Synthetic Population Resource for the routine dissection of complex traits. Genetics 2012, 191, 935–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, E.G.; Merkes, C.M.; McNeil, C.L.; Hoofer, S.R.; Sen, S.; Broman, K.W.; Long, A.D.; Macdonald, S.J. Genetic dissection of a model complex trait using the Drosophila Synthetic Population Resource. Genome Res. 2012, 22, 1558–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, E.G.; Sanderson, B.J.; McNeil, C.L.; Long, A.D.; Macdonald, S.J. Genetic dissection of the Drosophila melanogaster female head transcriptome reveals widespread allelic heterogeneity. PLoS Genet. 2014, 10, e1004322. [Google Scholar] [CrossRef] [Green Version]
- Ganguly-Fitzgerald, I.; Donlea, J.; Shaw, P.J. Waking experience affects sleep need in Drosophila. Science 2006, 313, 1775–1781. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 10 March 2020).
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar]
- DSPRqtl GitHub Site. Available online: https://github.com/egking/DSPRqtl (accessed on 5 March 2020).
- DSPR Website. Available online: www.FlyRILs.org (accessed on 5 March 2020).
- Perkins, L.A.; Holderbaum, L.; Tao, R.; Hu, Y.; Sopko, R.; McCall, K.; Yang-Zhou, D.; Flockhart, I.; Binari, R.; Shim, H.S.; et al. The Transgenic RNAi Project at Harvard Medical School: Resources and Validation. Genetics 2015, 201, 843–852. [Google Scholar] [CrossRef]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef]
- Huang, W.; Massouras, A.; Inoue, Y.; Peiffer, J.; Ramia, M.; Tarone, A.M.; Turlapati, L.; Zichner, T.; Zhu, D.; Lyman, R.F.; et al. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res. 2014, 24, 1193–1208. [Google Scholar] [CrossRef] [Green Version]
- King, E.G.; Long, A.D. The Beavis Effect in Next-Generation Mapping Panels in Drosophila melanogaster. G3 (Bethesda) 2017, 7, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, T.F.; Richards, S.; Stone, E.A.; Barbadilla, A.; Ayroles, J.F.; Zhu, D.; Casillas, S.; Han, Y.; Magwire, M.M.; Cridland, J.M.; et al. The Drosophila melanogaster Genetic Reference Panel. Nature 2012, 482, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; R Core Team. nlme: Linear and Nonlinear Mixed Effects Models, R package version 3.1-137; R Foundation: Vienna, Austria, 2018. [Google Scholar]
- Cloud-Richardson, K.M.; Smith, B.R.; Macdonald, S.J. Genetic dissection of intraspecific variation in a male-specific sexual trait in Drosophila melanogaster. Heredity (Edinb) 2016, 117, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, A.; Hermsen, R.; Guryev, V.; Stridh, P.; Graham, D.; McBride, M.W.; Foroud, T.; Calderari, S.; Diez, M.; Ockinger, J.; et al. Combined sequence-based and genetic mapping analysis of complex traits in outbred rats. Nat. Genet. 2013, 45, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.M.; Good, R.T.; Appleton, B.; Sherrard, J.; Raymant, G.C.; Bogwitz, M.R.; Martin, J.; Daborn, P.J.; Goddard, M.E.; Batterham, P.; et al. Copy number variation and transposable elements feature in recent, ongoing adaptation at the Cyp6g1 locus. PLoS Genet. 2010, 6, e1000998. [Google Scholar] [CrossRef] [Green Version]
- Hormozdiari, F.; Zhu, A.; Kichaev, G.; Ju, C.J.; Segre, A.V.; Joo, J.W.J.; Won, H.; Sankararaman, S.; Pasaniuc, B.; Shifman, S.; et al. Widespread Allelic Heterogeneity in Complex Traits. Am. J. Hum. Genet. 2017, 100, 789–802. [Google Scholar] [CrossRef] [Green Version]
- OMIM: Online Mendelian Inheritance in Man. Available online: https://omim.org/ (accessed on 5 March 2020).
- Darvasi, A. Experimental strategies for the genetic dissection of complex traits in animal models. Nat. Genet. 1998, 18, 19–24. [Google Scholar] [CrossRef]
- Darvasi, A.; Soller, M. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics 1995, 141, 1199–1207. [Google Scholar]
- Attrill, H.; Falls, K.; Goodman, J.L.; Millburn, G.H.; Antonazzo, G.; Rey, A.J.; Marygold, S.J. FlyBase: Establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acids Res. 2016, 44, D786–D792. [Google Scholar] [CrossRef] [Green Version]
- Gamazon, E.R.; Segre, A.V.; van de Bunt, M.; Wen, X.; Xi, H.S.; Hormozdiari, F.; Ongen, H.; Konkashbaev, A.; Derks, E.M.; Aguet, F.; et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 2018, 50, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Gilad, Y.; Rifkin, S.A.; Pritchard, J.K. Revealing the architecture of gene regulation: The promise of eQTL studies. Trends Genet. 2008, 24, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolae, D.L.; Gamazon, E.; Zhang, W.; Duan, S.; Dolan, M.E.; Cox, N.J. Trait-associated SNPs are more likely to be eQTLs: Annotation to enhance discovery from GWAS. PLoS Genet. 2010, 6, e1000888. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, H.; Kitamoto, T. The steroid molting hormone Ecdysone regulates sleep in adult Drosophila melanogaster. Genetics 2010, 185, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Long, A.D.; Macdonald, S.J.; King, E.G. Dissecting complex traits using the Drosophila Synthetic Population Resource. Trends Genet. 2014, 30, 488–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everman, E.R.; McNeil, C.L.; Hackett, J.L.; Bain, C.L.; Macdonald, S.J. Dissection of Complex, Fitness-Related Traits in Multiple Drosophila Mapping Populations Offers Insight into the Genetic Control of Stress Resistance. Genetics 2019, 211, 1449–1467. [Google Scholar] [CrossRef] [Green Version]
- Najarro, M.A.; Hackett, J.L.; Macdonald, S.J. Loci Contributing to Boric Acid Toxicity in Two Reference Populations of Drosophila melanogaster. G3 (Bethesda) 2017, 7, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Najarro, M.A.; Hackett, J.L.; Smith, B.R.; Highfill, C.A.; King, E.G.; Long, A.D.; Macdonald, S.J. Identifying Loci Contributing to Natural Variation in Xenobiotic Resistance in Drosophila. PLoS Genet. 2015, 11, e1005663. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, A.; Price, J.L.; Man, B.; Young, M.W. Loss of circadian behavioral rhythms and per RNA oscillations in the Drosophila mutant timeless. Science 1994, 263, 1603–1606. [Google Scholar] [CrossRef]
- Vosshall, L.B.; Price, J.L.; Sehgal, A.; Saez, L.; Young, M.W. Block in nuclear localization of period protein by a second clock mutation, timeless. Science 1994, 263, 1606–1609. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, M.S.; Tempel, B.L. Genetic dissection of monoamine neurotransmitter synthesis in Drosophila. Nature 1983, 303, 67–70. [Google Scholar] [CrossRef]
- Jordan, K.W.; Morgan, T.J.; Mackay, T.F. Quantitative trait loci for locomotor behavior in Drosophila melanogaster. Genetics 2006, 174, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepson, J.E.; Shahidullah, M.; Lamaze, A.; Peterson, D.; Pan, H.; Koh, K. dyschronic, a Drosophila homolog of a deaf-blindness gene, regulates circadian output and Slowpoke channels. PLoS Genet. 2012, 8, e1002671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, J.E.; Raizen, D.M.; Maycock, M.H.; Maislin, G.; Pack, A.I. A video method to study Drosophila sleep. Sleep 2008, 31, 1587–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbe, D.S.; Bollinger, W.L.; Vigderman, A.; Masek, P.; Gertowski, J.; Sehgal, A.; Keene, A.C. Context-specific comparison of sleep acquisition systems in Drosophila. Biol. Open 2015, 4, 1558–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donelson, N.C.; Kim, E.Z.; Slawson, J.B.; Vecsey, C.G.; Huber, R.; Griffith, L.C. High-resolution positional tracking for long-term analysis of Drosophila sleep and locomotion using the “tracker” program. PLoS ONE 2012, 7, e37250. [Google Scholar] [CrossRef]
- Faville, R.; Kottler, B.; Goodhill, G.J.; Shaw, P.J.; van Swinderen, B. How deeply does your mutant sleep? Probing arousal to better understand sleep defects in Drosophila. Sci. Rep. 2015, 5, 8454. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTL Group | Phenotypes a | Chr | Interval Size (Mb) b | Num Genes c | Num Local eQTL d |
|---|---|---|---|---|---|
| QG1 | DBN, DBL | X | 0.70 | 57 | 26 |
| QG2 | DST | 2L | 1.91 | 240 | 74 |
| QG3 | DBN | 2L | 0.60 | 66 | 18 |
| QG4 | LBN | 2L | 2.03 | 158 | 50 |
| QG5 | LWA, LBN | 2L | 1.17 | 74 | 16 |
| QG6 | LST | 2L | 0.75 | 103 | 52 |
| QG7 | DST, DBN, LBL, DBL | 2c e | 7.92 | 316 e | 178 e |
| QG8 | DBL | 3L | 0.90 | 85 | 30 |
| QG9 | LBL | 3L | 0.27 | 35 | 17 |
| QG10 | LST, DST, LBN, DBL | 3L | 0.64 | 48 | 23 |
| QG11 | LWA, DWA, LBN | 3R | 1.21 | 124 | 41 |
| QG12 | DWA | 3R | 0.85 | 84 | 27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
R. Smith, B.; J. Macdonald, S. Dissecting the Genetic Basis of Variation in Drosophila Sleep Using a Multiparental QTL Mapping Resource. Genes 2020, 11, 294. https://doi.org/10.3390/genes11030294
R. Smith B, J. Macdonald S. Dissecting the Genetic Basis of Variation in Drosophila Sleep Using a Multiparental QTL Mapping Resource. Genes. 2020; 11(3):294. https://doi.org/10.3390/genes11030294
Chicago/Turabian StyleR. Smith, Brittny, and Stuart J. Macdonald. 2020. "Dissecting the Genetic Basis of Variation in Drosophila Sleep Using a Multiparental QTL Mapping Resource" Genes 11, no. 3: 294. https://doi.org/10.3390/genes11030294
APA StyleR. Smith, B., & J. Macdonald, S. (2020). Dissecting the Genetic Basis of Variation in Drosophila Sleep Using a Multiparental QTL Mapping Resource. Genes, 11(3), 294. https://doi.org/10.3390/genes11030294