Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells



Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Drug Treatments
2.3. RNA Isolation and Quantitative Reverse Transcriptase Polymerase Chain Reaction (RT-qPCR)
2.4. DNA Isolation and PCR
2.5. Chromatin Immunoprecipitation (ChIP)
2.6. Western Blot Analyses
2.7. Immunostaining
2.8. Statistical Analysis
3. Results
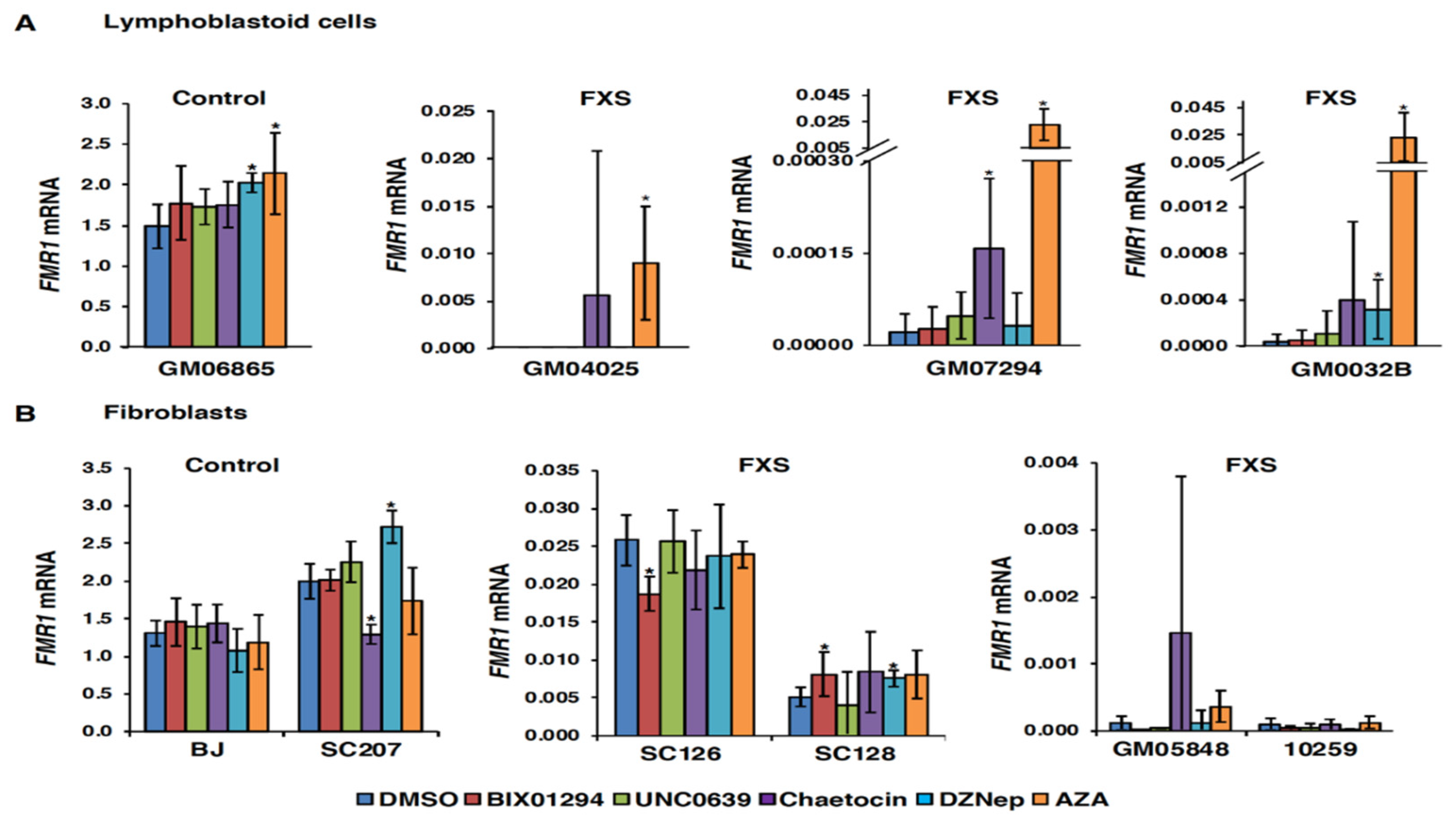
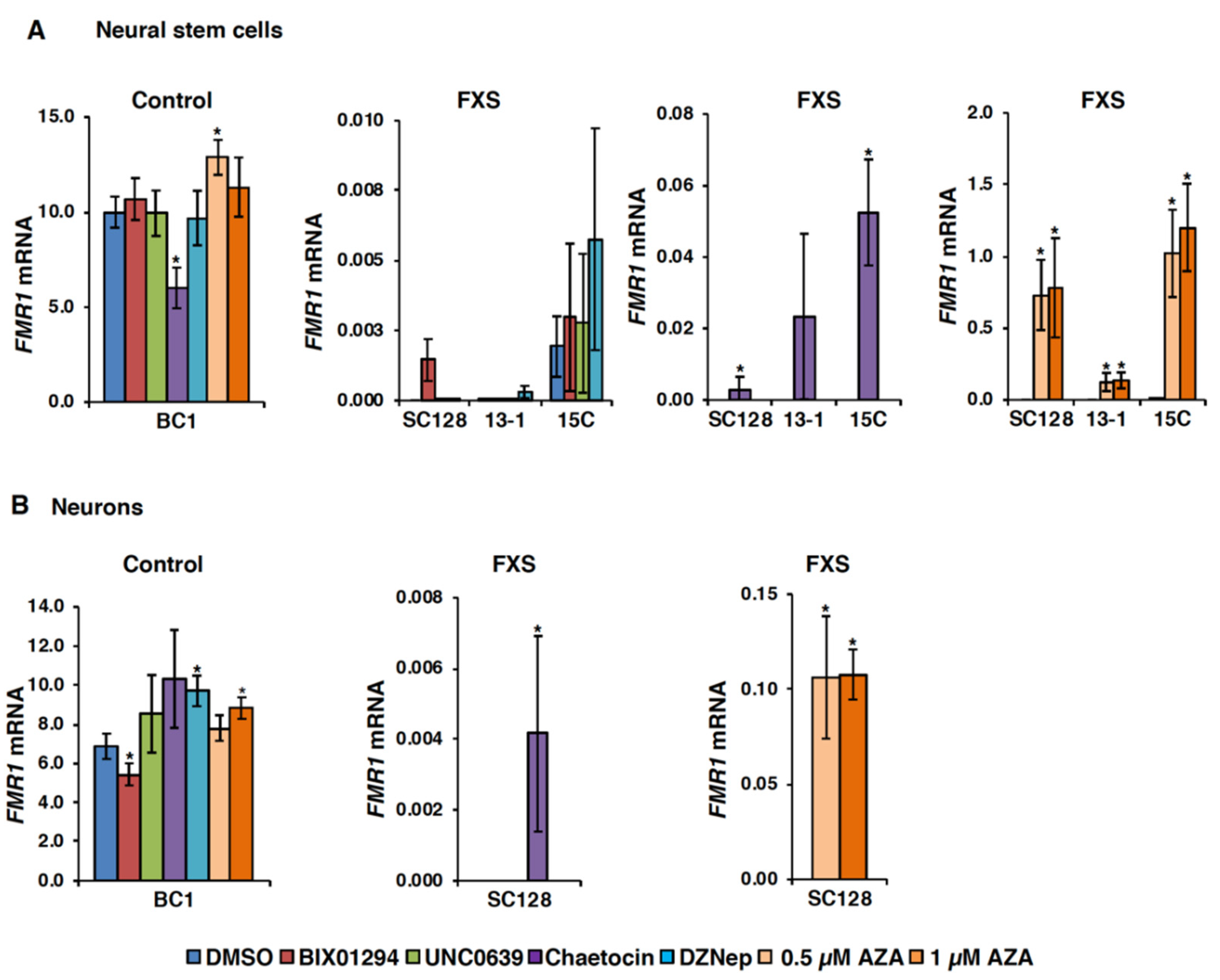
3.1. Small Molecules Targeting H3K9 Methylation, by Themselves, are not Very Effective at Activating FMR1 Expression in FXS-Patient-Derived Cells
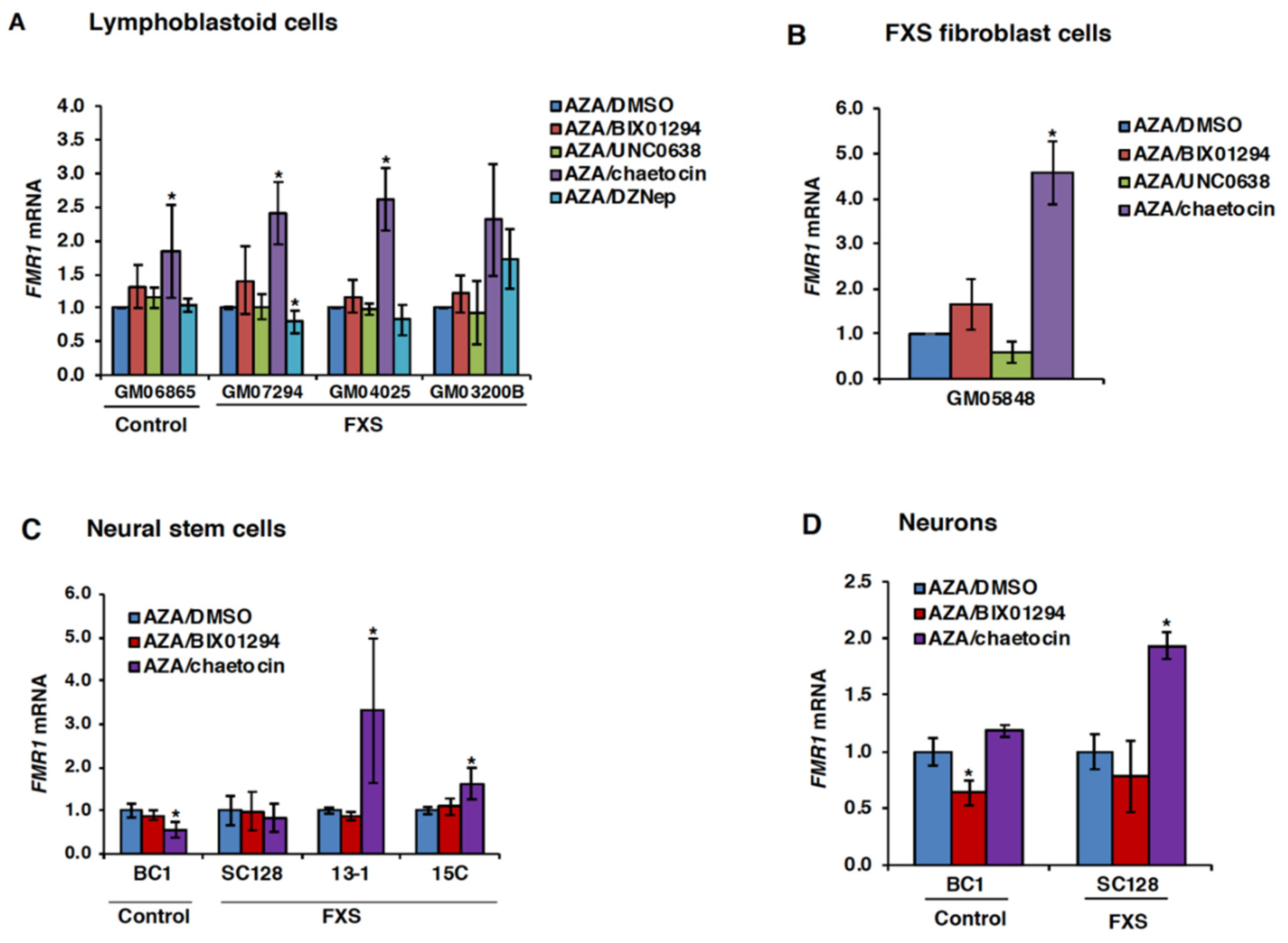
3.2. Chaetocin Potentiates the Expression of AZA-Reactivated FM Alleles in FXS Patient Cells
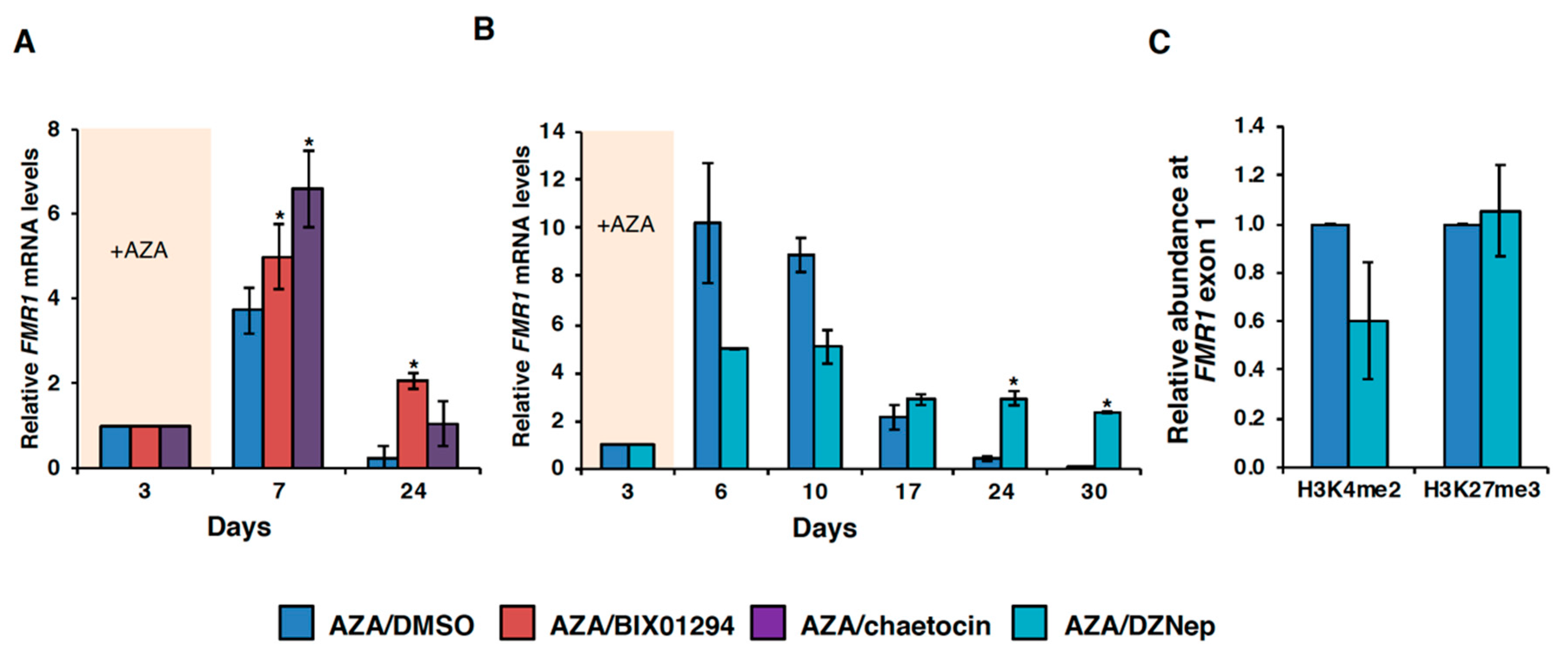
3.3. HMT Inhibitors Delay Re-Silencing of AZA-Reactivated Alleles in FXS Lymphoblastoid Cells
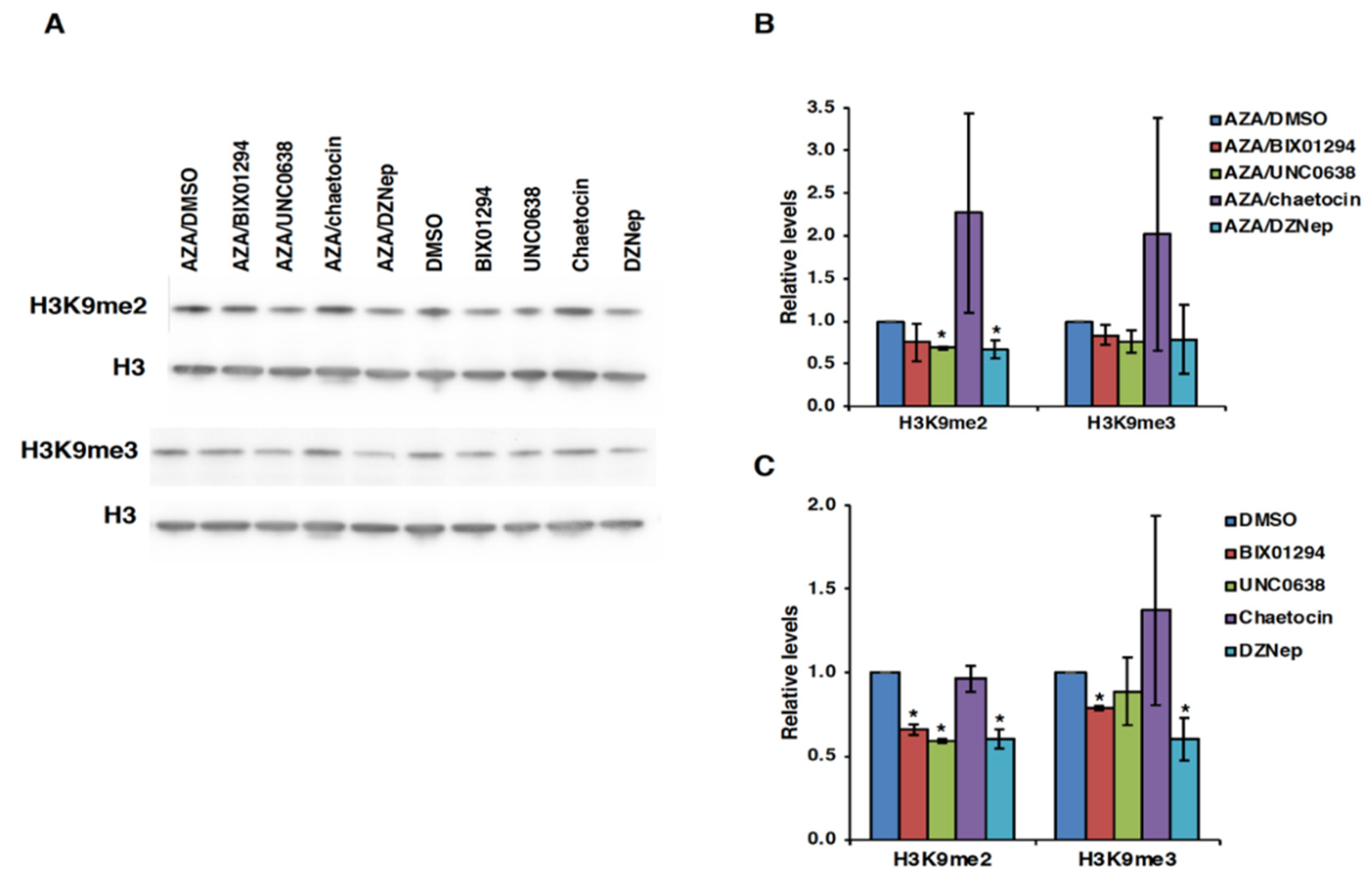
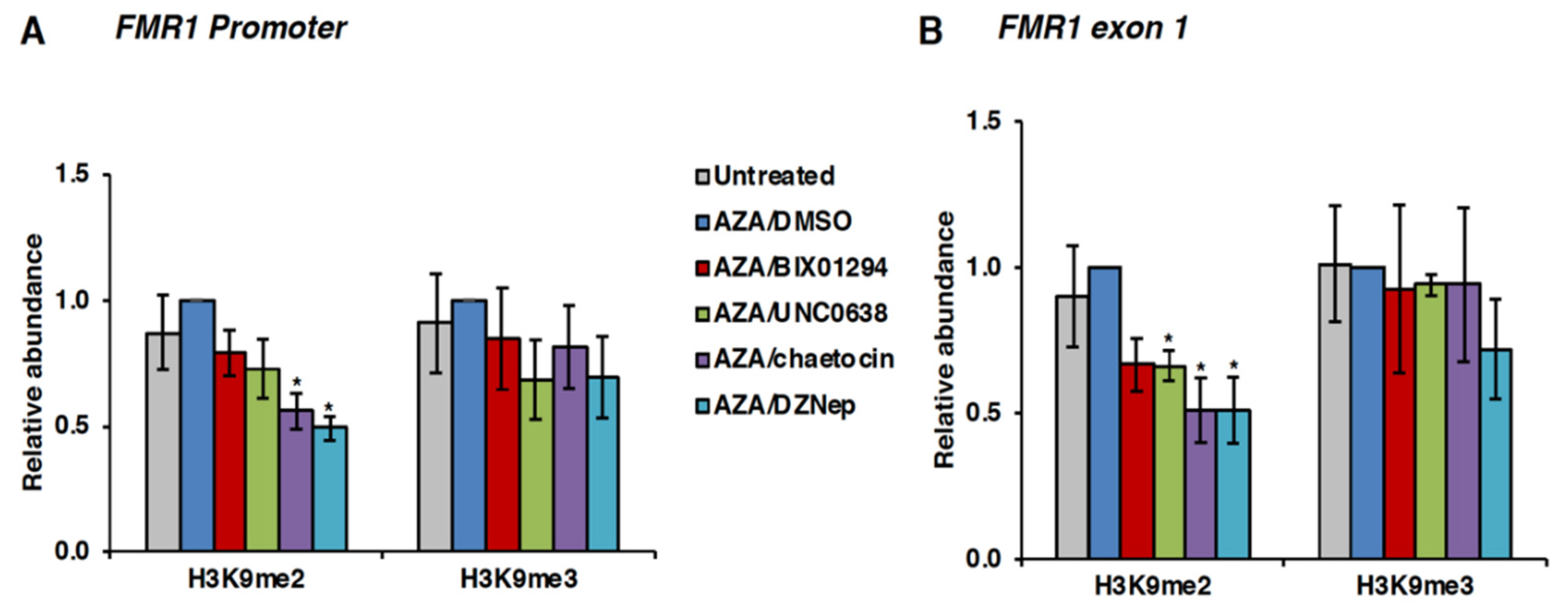
3.4. Effect of HMT Inhibitors on H3K9 Methylation of the FMR1 Gene in FXS Patient Cells
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X syndrome: A review of associated medical problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef]
- Coffee, B.; Zhang, F.; Warren, S.T.; Reines, D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999, 22, 98–101. [Google Scholar] [CrossRef]
- Coffee, B.; Zhang, F.; Ceman, S.; Warren, S.T.; Reines, D. Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile X syndrome. Am. J. Hum. Genet. 2002, 71, 923–932. [Google Scholar] [CrossRef]
- Kumari, D.; Usdin, K. The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum. Mol. Genet. 2010, 19, 4634–4642. [Google Scholar] [CrossRef]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef]
- Kumari, D.; Usdin, K. Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet. 2014, 23, 6575–6583. [Google Scholar] [CrossRef]
- Hecht, M.; Tabib, A.; Kahan, T.; Orlanski, S.; Gropp, M.; Tabach, Y.; Yanuka, O.; Benvenisty, N.; Keshet, I.; Cedar, H. Epigenetic mechanism of FMR1 inactivation in Fragile X syndrome. Int. J. Dev. Biol. 2017, 61, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 2016, 25, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, R.; Tabolacci, E.; Zalfa, F.; Zito, I.; Terracciano, A.; Moscato, U.; Bagni, C.; Oostra, B.; Chiurazzi, P.; Neri, G. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 2005, 14, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Moscato, U.; Zalfa, F.; Bagni, C.; Chiurazzi, P.; Neri, G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008, 16, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef]
- Peters, A.H.; Kubicek, S.; Mechtler, K.; O’Sullivan, R.J.; Derijck, A.A.; Perez-Burgos, L.; Kohlmaier, A.; Opravil, S.; Tachibana, M.; Shinkai, Y.; et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 2003, 12, 1577–1589. [Google Scholar] [CrossRef]
- Greiner, D.; Bonaldi, T.; Eskeland, R.; Roemer, E.; Imhof, A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat. Chem. Biol. 2005, 1, 143–145. [Google Scholar] [CrossRef]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef]
- Chang, Y.; Zhang, X.; Horton, J.R.; Upadhyay, A.K.; Spannhoff, A.; Liu, J.; Snyder, J.P.; Bedford, M.T.; Cheng, X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. [Google Scholar] [CrossRef]
- Vedadi, M.; Barsyte-Lovejoy, D.; Liu, F.; Rival-Gervier, S.; Allali-Hassani, A.; Labrie, V.; Wigle, T.J.; Dimaggio, P.A.; Wasney, G.A.; Siarheyeva, A.; et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7, 566–574. [Google Scholar] [CrossRef]
- Glazer, R.I.; Hartman, K.D.; Knode, M.C.; Richard, M.M.; Chiang, P.K.; Tseng, C.K.; Marquez, V.E. 3-Deazaneplanocin: A new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem. Biophys. Res. Commun. 1986, 135, 688–694. [Google Scholar] [CrossRef]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kim, K.C. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem. Biophys. Res. Commun. 2013, 438, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Bhattacharya, A.; Nadel, J.; Moulton, K.; Zeak, N.M.; Glicksman, A.; Dobkin, C.; Brick, D.J.; Schwartz, P.H.; Smith, C.B.; et al. Identification of fragile X syndrome specific molecular markers in human fibroblasts: A useful model to test the efficacy of therapeutic drugs. Hum. Mutat. 2014, 35, 1485–1494. [Google Scholar] [CrossRef]
- Kumari, D.; Swaroop, M.; Southall, N.; Huang, W.; Zheng, W.; Usdin, K. High-throughput screening to identify compounds that increase fragile X mental retardation protein expression in neural stem cells differentiated from fragile X syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl. Med. 2015, 4, 800–808. [Google Scholar] [CrossRef]
- Beers, J.; Linask, K.L.; Chen, J.A.; Siniscalchi, L.I.; Lin, Y.; Zheng, W.; Rao, M.; Chen, G. A cost-effective and efficient reprogramming platform for large-scale production of integration-free human induced pluripotent stem cells in chemically defined culture. Sci. Rep. 2015, 5, 11319. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Hayward, B.E.; Zhou, Y.; Kumari, D.; Usdin, K. A set of assays for the comprehensive analysis of FMR1 alleles in the fragile X-related disorders. J. Mol. Diagn. 2016, 18, 762–774. [Google Scholar] [CrossRef]
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism 2016, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Pomponi, M.G.; Willemsen, R.; Oostra, B.A.; Neri, G. In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 1998, 7, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Biacsi, R.; Kumari, D.; Usdin, K. SIRT1 inhibition alleviates gene silencing in Fragile X mental retardation syndrome. PLoS Genet. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, R.; Pomponi, M.G.; Tabolacci, E.; Oostra, B.; Chiurazzi, P.; Neri, G. Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res. 2002, 30, 3278–3285. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Lakshmikuttyamma, A.; Scott, S.A.; DeCoteau, J.F.; Geyer, C.R. Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene 2010, 29, 576–588. [Google Scholar] [CrossRef]
- Chen, P.; Yao, J.F.; Huang, R.F.; Zheng, F.F.; Jiang, X.H.; Chen, X.; Chen, J.; Li, M.; Huang, H.F.; Jiang, Y.P.; et al. Effect of BIX-01294 on H3K9me2 levels and the imprinted gene Snrpn in mouse embryonic fibroblast cells. Biosci. Rep. 2015, 35, e00257. [Google Scholar] [CrossRef]
- Chiba, T.; Saito, T.; Yuki, K.; Zen, Y.; Koide, S.; Kanogawa, N.; Motoyama, T.; Ogasawara, S.; Suzuki, E.; Ooka, Y.; et al. Histone lysine methyltransferase SUV39H1 is a potent target for epigenetic therapy of hepatocellular carcinoma. Int. J. Cancer 2015, 136, 289–298. [Google Scholar] [CrossRef]
- Renneville, A.; Van Galen, P.; Canver, M.C.; McConkey, M.; Krill-Burger, J.M.; Dorfman, D.M.; Holson, E.B.; Bernstein, B.E.; Orkin, S.H.; Bauer, D.E.; et al. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood 2015, 126, 1930–1939. [Google Scholar] [CrossRef]
- Song, X.; Zhao, Z.; Qi, X.; Tang, S.; Wang, Q.; Zhu, T.; Gu, Q.; Liu, M.; Li, J. Identification of epipolythiodioxopiperazines HDN-1 and chaetocin as novel inhibitor of heat shock protein 90. Oncotarget 2015, 6, 5263–5274. [Google Scholar] [CrossRef]
- Liu, X.R.; Zhou, L.H.; Hu, J.X.; Liu, L.M.; Wan, H.P.; Zhang, X.Q. UNC0638, a G9a inhibitor, suppresses epithelialmesenchymal transitionmediated cellular migration and invasion in triple negative breast cancer. Mol. Med. Rep. 2018, 17, 2239–2244. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, Z.; Cedar, H. DNA methylation: A molecular lock. Curr. Biol. 1997, 7, R305–R307. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Pomponi, M.G.; Pietrobono, R.; Bakker, C.E.; Neri, G.; Oostra, B.A. Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 1999, 8, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Soragni, E.; Jenssen, K.; Burnett, R.; Herman, D.; Coppola, G.; Geschwind, D.H.; Gottesfeld, J.M.; Pandolfo, M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE 2008, 3, e1958. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Mancano, G.; Lanni, S.; Palumbo, F.; Goracci, M.; Ferre, F.; Helmer-Citterich, M.; Neri, G. Genome-wide methylation analysis demonstrates that 5-aza-2-deoxycytidine treatment does not cause random DNA demethylation in fragile X syndrome cells. Epigenetics Chromatin 2016, 9, 12. [Google Scholar] [CrossRef]
- Vershkov, D.; Fainstein, N.; Suissa, S.; Golan-Lev, T.; Ben-Hur, T.; Benvenisty, N. FMR1 Reactivating treatments in fragile X iPSC-derived neural progenitors In Vitro and In Vivo. Cell Rep. 2019, 26, 2531–2539. [Google Scholar] [CrossRef]
- Kim, K.; Hessl, D.; Randol, J.L.; Espinal, G.M.; Schneider, A.; Protic, D.; Aydin, E.Y.; Hagerman, R.J.; Hagerman, P.J. Association between IQ and FMR1 protein (FMRP) across the spectrum of CGG repeat expansions. PLoS ONE 2019, 14, e0226811. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, F.; Lokey, L.K.; Chastain, J.L.; Lakkis, L.; Eberhart, D.; Warren, S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science 1995, 268, 731–734. [Google Scholar] [CrossRef]
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001, 10, 1449–1454. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef]
- Sellier, C.; Freyermuth, F.; Tabet, R.; Tran, T.; He, F.; Ruffenach, F.; Alunni, V.; Moine, H.; Thibault, C.; Page, A.; et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, 3, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.M.; Wright, S.E.; Kearse, M.G.; Haenfler, J.M.; Flores, B.N.; Liu, Y.; Ifrim, M.F.; Glineburg, M.R.; Krans, A.; Jafar-Nejad, P.; et al. A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat. Neurosci. 2020, 23, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Sheth, U.J.; Flores, B.N.; Wright, S.E.; Sutter, A.B.; Kearse, M.G.; Barmada, S.J.; Ivanova, M.I.; Todd, P.K. High-throughput screening yields several small-molecule inhibitors of repeat-associated non-AUG translation. J. Biol. Chem. 2019, 294, 18624–18638. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line Name | Cell Type | FXS/Control | CGG-Repeat Size/MethyLation |
|---|---|---|---|
| BJ | fibroblast | Control | 26 |
| SC207 | fibroblast | Control | 20 |
| SC126 | fibroblast | FXS | Mosaic for PM/ methylated FM |
| SC128 | fibroblast | FXS | Mosaic for PM/ methylated FM |
| C10259 | fibroblast | FXS | Methylated FM (1363 by SB) |
| C10700 | fibroblast | FXS | Mosaic for PM/ methylated FM |
| GM05848 | fibroblast | FXS | Methylated FM (~700) |
| GM06865 | lymphoblastoid | Control | 30 |
| GM0032B | lymphoblastoid | FXS | Methylated FM (570) |
| GM04025 | lymphoblastoid | FXS | Methylated FM (645) |
| GM07294 | lymphoblastoid | FXS | Methylated FM (745) |
| 13-1 | iPSCs | FXS | Methylated FM (463) |
| 15C | iPSCs | FXS | Methylated FM (330) |
| SC128 | iPSCs | FXS | Methylated FM (300) |
| BC1 NSC | NSC | Control | NA |
| SC128 NSC | NSC | FXS | Methylated FM (300) |
| SC128 neurons | neurons | FXS | Methylated FM (300) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumari, D.; Sciascia, N.; Usdin, K. Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells. Genes 2020, 11, 356. https://doi.org/10.3390/genes11040356
Kumari D, Sciascia N, Usdin K. Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells. Genes. 2020; 11(4):356. https://doi.org/10.3390/genes11040356
Chicago/Turabian StyleKumari, Daman, Nicholas Sciascia, and Karen Usdin. 2020. "Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells" Genes 11, no. 4: 356. https://doi.org/10.3390/genes11040356
APA StyleKumari, D., Sciascia, N., & Usdin, K. (2020). Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells. Genes, 11(4), 356. https://doi.org/10.3390/genes11040356