The Emerging Role of the RBM20 and PTBP1 Ribonucleoproteins in Heart Development and Cardiovascular Diseases

 , and
, and 
Abstract
:1. Introduction
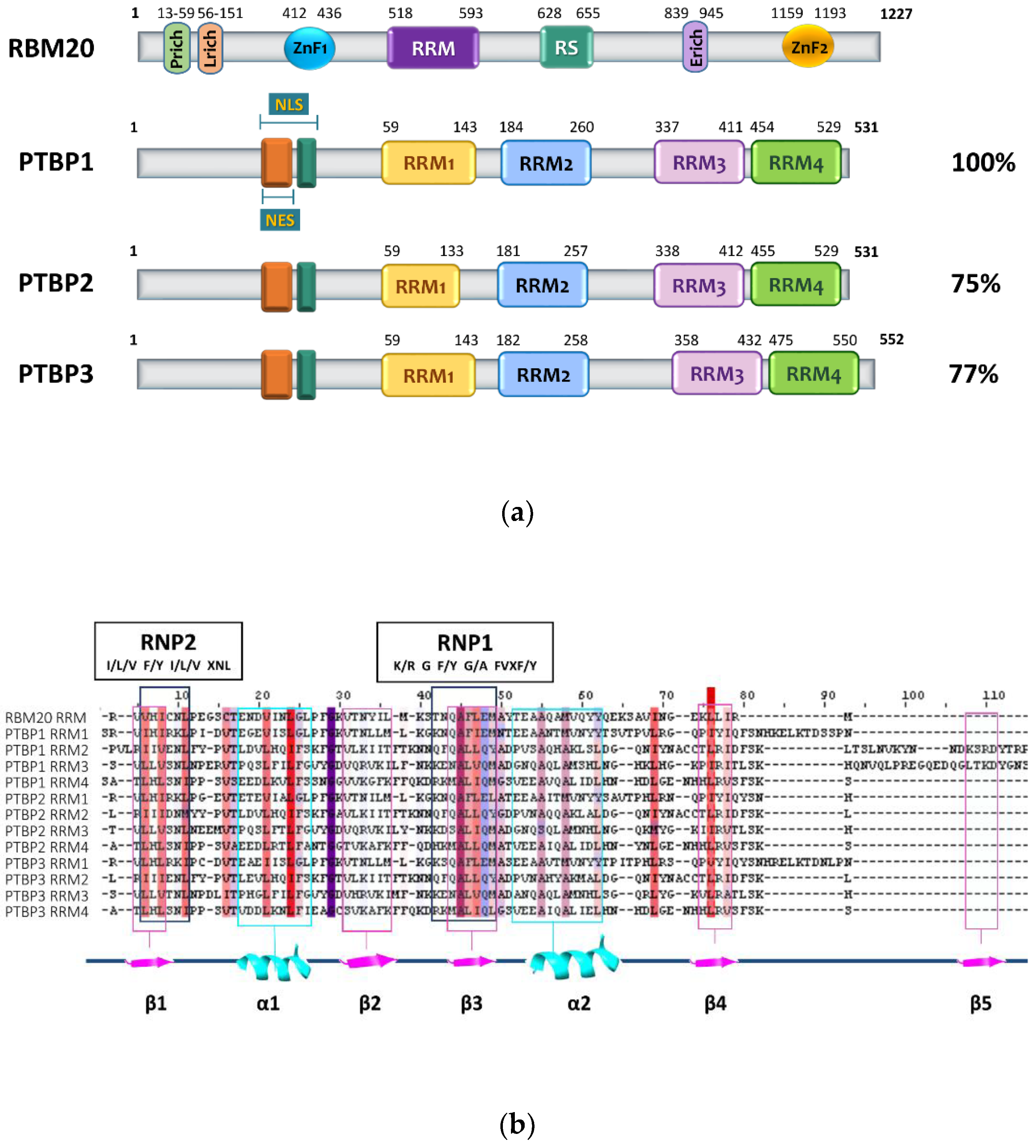
2. RBM20 Protein Structure
3. PTBP Proteins’ Structure and Function
4. Regulation of Alternative Splicing Events in Heart by RBM20 and PTB
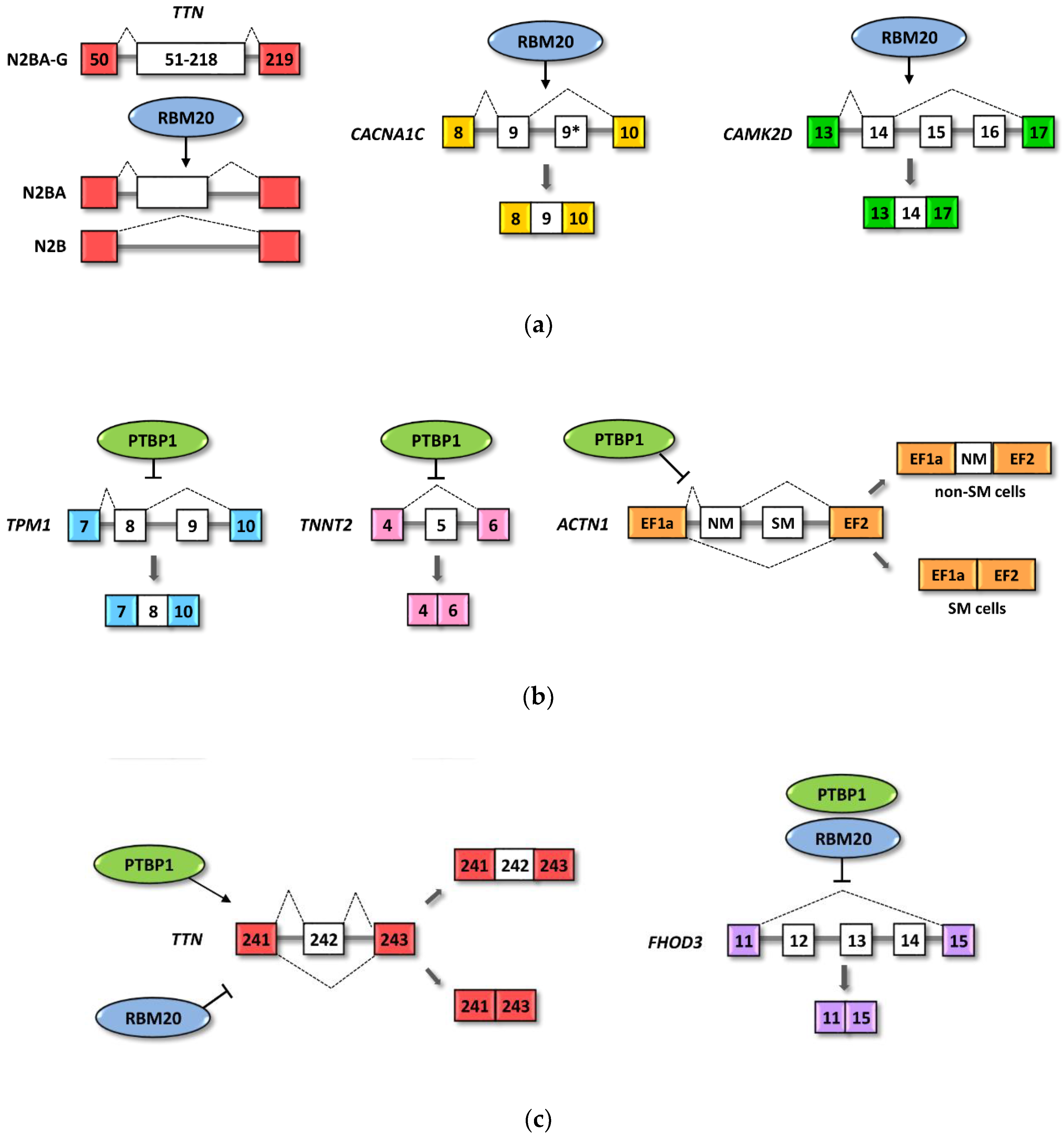
4.1. RBM20 Regulated Cardiac Pre-mRNAs
4.2. Role of RBM20 in Heart Diseases
4.3. PTBP1 Regulated Heart Pre-mRNA
4.4. RBM20 and PTBP1 Combinatorial Effects on Alternative Splicing
4.5. RBM20 and RBM24 Cooperation in Alternative Splicing
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Rath, E.; Bai, Y. Comparison of Alternative Splicing Junction Detection Tools Using RNASeq Data. Curr. Genom. 2017, 18, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, K.; Takeda, J.I.; Masuda, A. Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdiscip. Rev. RNA 2018, 9, e1451. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Darnell, R.B. RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet. 2010, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.D.; Barrios-Rodiles, M.; Çolak, R.; Irimia, M.; Kim, T.H.; Calarco, J.A.; Wang, X.; Pan, Q.; O’Hanlon, D.; Kim, P.M.; et al. Tissue-Specific Alternative Splicing Remodels Protein-Protein Interaction Networks. Mol. Cell 2012, 46, 884–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Çolak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Traunmüller, L.; Gomez, A.M.; Nguyen, T.M.; Scheiffele, P. Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science 2016, 352, 982–986. [Google Scholar] [CrossRef]
- Hinkle, E.R.; Wiedner, H.J.; Black, A.J.; Giudice, J. RNA processing in skeletal muscle biology and disease. Transcription 2019, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Nikonova, E.; Kao, S.-Y.; Spletter, M.L. Contributions of alternative splicing to muscle type development and function. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.-M.; Li, J.; Huang, L.-F.; Lin, J.; Zhang, J.; Min, Q.-H.; Yang, W.-M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenasa, H.; Hertel, K.J. Combinatorial regulation of alternative splicing. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194392. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Aspedia. Available online: http://combio.snu.ac.kr/aspedia/ (accessed on 2 April 2020).
- Hyung, D.; Kim, J.; Cho, S.Y.; Park, C. ASpedia: A comprehensive encyclopedia of human alternative splicing. Nucleic Acids Res. 2018, 46, D58–D63. [Google Scholar] [CrossRef]
- SpliceAdF. Available online: http://srv00.recas.ba.infn.it/SpliceAidF/ (accessed on 2 April 2020).
- Giulietti, M.; Piva, F.; D’Antonio, M.; De Meo, P.D.O.; Paoletti, D.; Castrignanò, T.; D’Erchia, A.M.; Picardi, E.; Zambelli, F.; Principato, G.; et al. SpliceAid-F: A database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 2013, 41, 125–131. [Google Scholar] [CrossRef]
- MiasDB. Available online: http://47.88.84.236/Miasdb/ (accessed on 2 April 2020).
- Xing, Y.; Zhao, X.; Yu, T.; Liang, D.; Li, J.; Wei, G.; Liu, G.; Cui, X.; Zhao, H.; Cai, L. MiasDB: A database of molecular interactions associated with alternative splicing of human pre-mRNAs. PLoS ONE 2016, 11, e0155443. [Google Scholar] [CrossRef] [Green Version]
- SplicePort. Available online: https://spliceport.cbcb.umd.edu/ (accessed on 2 April 2020).
- Dogan, R.I.; Getoor, L.; Wilbur, W.J.; Mount, S.M. SplicePort-An interactive splice-site analysis tool. Nucleic Acids Res. 2007, 35, 285–291. [Google Scholar] [CrossRef]
- HSF. Available online: http://www.umd.be/HSF/ (accessed on 2 April 2020).
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, Y.; Li, X.; Chen, G.; Zhong, L.; Chen, G.; Liao, Y.; Liao, W.; Bin, J. Genome-wide analysis of alternative splicing during human heart development. Sci. Rep. 2016, 6, 35520. [Google Scholar] [CrossRef] [Green Version]
- Beqqali, A. Alternative splicing in cardiomyopathy. Biophys. Rev. 2018, 10, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Lu, L.; Guo, W.; Ren, J.; Yang, J. Emerging Role for RBM20 and its Splicing Substrates in Cardiac Function and Heart Failure. Curr. Pharm. Des. 2016, 22, 4744–4751. [Google Scholar] [CrossRef] [PubMed]
- Weeland, C.J.; Van den Hoogenhof, M.M.; Beqqali, A.; Creemers, E.E. Insights into alternative splicing of sarcomeric genes in the heart. J. Mol. Cell. Cardiol. 2015, 81, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kimura, A.; Kuroyanagi, H. Alternative splicing regulator RBM20 and cardiomyopathy. Front. Mol. Biosci. 2018, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Rexiati, M.; Sun, M.; Guo, W. Muscle-specific Mis-splicing and heart disease exemplified by RBM20. Genes 2018, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; De Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in Ribonucleic Acid Binding Protein Gene Cause Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Dauksaite, V.; Gotthardt, M. Molecular basis of titin exon exclusion by RBM20 and the novel titin splice regulator PTB4. Nucleic Acids Res. 2018, 46, 5227–5238. [Google Scholar] [CrossRef]
- Lorenzi, P.; Sangalli, A.; Fochi, S.; Dal Molin, A.; Malerba, G.; Zipeto, D.; Romanelli, M.G. RNA-binding proteins RBM20 and PTBP1 regulate the alternative splicing of FHOD3. Int. J. Biochem. Cell Biol. 2019, 106, 74–83. [Google Scholar] [CrossRef]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Filippello, A.; Lorenzi, P.; Bergamo, E.; Romanelli, M.G. Identification of nuclear retention domains in the RBM20 protein. FEBS Lett. 2013, 587, 2989–2995. [Google Scholar] [CrossRef] [Green Version]
- Murayama, R.; Kimura-Asami, M.; Togo-Ohno, M.; Yamasaki-Kato, Y.; Naruse, T.K.; Yamamoto, T.; Hayashi, T.; Ai, T.; Spoonamore, K.G.; Kovacs, R.J.; et al. Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-Binding Motif Protein 20 (RBM20) through nuclear localization. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zahr, H.C.; Jaalouk, D.E. Exploring the crosstalk between LMNA and splicing machinery gene mutations in Dilated Cardiomyopathy. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.; Martin, D.; Clamp, M.; Barton, G. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksaite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liss, M.; Radke, M.H.; Eckhard, J.; Neuenschwander, M.; Dauksaite, V.; Von Kries, J.P.; Gotthardt, M. Drug discovery with an RBM20 dependent titin splice reporter identifies cardenolides as lead structures to improve cardiac filling. PLoS ONE 2018, 13, e0198492. [Google Scholar] [CrossRef]
- Romanelli, M.G.; Diani, E.; Lievens, P.M.J. New insights into functional roles of the polypyrimidine tract-binding protein. Int. J. Mol. Sci. 2013, 14, 22906–22932. [Google Scholar] [CrossRef] [Green Version]
- Ghetti, A.; Piñol-Roma, S.; Michael, W.M.; Morandi, C.; Dreyfuss, G. hnRNP I, the polypyrimidine tract-binding protein: Distinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992, 20, 3671–3678. [Google Scholar] [CrossRef]
- Romanelli, M.G.; Weighardt, F.; Biamonti, G.; Riva, S.; Morandi, C. Sequence determinants for hnRNP I protein nuclear localization. Exp. Cell Res. 1997, 235, 300–304. [Google Scholar] [CrossRef]
- Romanelli, M.G.; Lorenzi, P.; Morandi, C. Organization of the human gene encoding heterogeneous nuclear ribonucleoprotein type I (hnRNP I) and characterization of hnRNP I related pseudogene. Gene 2000, 255, 267–272. [Google Scholar] [CrossRef]
- Oberstrass, F.C.; Auwetor, S.D.; Erat, M.; Hargous, Y.; Henning, A.; Wenter, P.; Reymond, L.; Amir-Ahmady, B.; Pitsch, S.; Black, D.L.; et al. Structural biology—Structure of PTB bound to RNA: Specific binding and implications for splicing regulation. Science 2005, 309, 2054–2057. [Google Scholar] [CrossRef]
- Romanelli, M.G.; Faggioli, L.; Lorenzi, P.; Morandi, C. Cloning and functional characterization of the human heterogeneous nuclear ribonucleoprotein type I promoter. Biochim. Biophys. Acta 2001, 1520, 85–88. [Google Scholar] [CrossRef]
- Wollerton, M.C.; Gooding, C.; Wagner, E.J.; Garcia-Blanco, M.A.; Smith, C.W.J. Autoregulation of Polypyrimidine Tract Binding Protein by Alternative Splicing Leading to Nonsense-Mediated Decay. Mol. Cell 2004, 13, 91–100. [Google Scholar] [CrossRef]
- Sawicka, K.; Bushell, M.; Spriggs, K.A.; Willis, A.E. Polypyrimidine-tract-binding protein: A multifunctional RNA-binding protein. Biochem. Soc. Trans. 2008, 36, 641–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Bahi, N.; Llovera, M.; Comella, J.X.; Sanchis, D. Polypyrimidine tract binding proteins (PTB) regulate the expression of apoptotic genes and susceptibility to caspase-dependent apoptosis in differentiating cardiomyocytes. Cell Death Differ. 2009, 16, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, M.H.; Wu, X.; Kodani, A.; Fan, J.; Doan, R.; Ozawa, M.; Ma, J.; Yoshida, N.; Reiter, J.F.; et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 2016, 166, 1147–1162.e15. [Google Scholar] [CrossRef] [Green Version]
- Monzón-Casanova, E.; Screen, M.; Díaz-Muñoz, M.D.; Coulson, R.M.R.; Bell, S.E.; Lamers, G.; Solimena, M.; Smith, C.W.J.; Turner, M. The RNA-binding protein PTBP1 is necessary for B cell selection in germinal centers article. Nat. Immunol. 2018, 19, 267–278. [Google Scholar] [CrossRef]
- Xue, Y.; Ouyang, K.; Huang, J.; Zhou, Y.; Ouyang, H.; Li, H.; Wang, G.; Wu, Q.; Wei, C.; Bi, Y.; et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated MicroRNA circuits. Cell 2013, 152, 82–96. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Nishimura, J.; Kagawa, Y.; Kano, Y.; Takahashi, Y.; Wu, X.; Hiraki, M.; Hamabe, A.; Konno, M.; Haraguchi, N.; et al. Significance of polypyrimidine tract-binding protein 1 expression in colorectal cancer. Mol. Cancer Ther. 2015, 14, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Chen, X.; Chen, Z.; Huang, M.; Dong, W.; Gu, P.; Zhang, J.; Zhou, Q.; Dong, W.; Han, J.; et al. Polypyrimidine tract binding protein 1 promotes lymphatic metastasis and proliferation of bladder cancer via alternative splicing of MEIS2 and PKM. Cancer Lett. 2019, 449, 31–44. [Google Scholar] [CrossRef]
- Keppetipola, N.; Sharma, S.; Li, Q.; Black, D.L. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 360–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanelli, M.G.; Lorenzi, P.; Morandi, C. Identification and analysis of the human neural polypyrimidine tract binding protein (nPTB) gene promoter region. Gene 2005, 356, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.P.; Chhabra, R.; Merran, J.D.; Schaughency, P.M.; Wheelan, S.J.; Corden, J.L.; Wong, P.C. PTBP1 and PTBP2 Repress Nonconserved Cryptic Exons. Cell Rep. 2016, 17, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zheng, S.; Han, A.; Lin, C.H.; Stoilov, P.; Fu, X.D.; Black, D.L. The splicing regulator PTBP2 controls a program of embryonic splicing required for neuronal maturation. Elife 2014, 2014. [Google Scholar] [CrossRef]
- Makeyev, E.V.; Zhang, J.; Carrasco, M.A.; Maniatis, T. The MicroRNA miR-124 Promotes Neuronal Differentiation by Triggering Brain-Specific Alternative Pre-mRNA Splicing. Mol. Cell 2007, 27, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Boutz, P.L.; Stoilov, P.; Li, Q.; Lin, C.H.; Chawla, G.; Ostrow, K.; Shiue, L.; Ares, M.; Black, D.L. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007, 21, 1636–1652. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, M.G.; Morandi, C. Importin α binds to an unusual bipartite nuclear localization signal in the heterogeneous ribonucleoprotein type I. Eur. J. Biochem. 2002, 269, 2727–2734. [Google Scholar] [CrossRef]
- Li, B.; Benedict Yen, T.S. Characterization of the nuclear export signal of polypyrimidine tract-binding protein. J. Biol. Chem. 2002, 277, 10306–10314. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.R.; Grüne, T.; Ghuman, J.; Kelly, G.; Ladas, A.; Matthews, S.; Curry, S. Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. EMBO J. 2000, 19, 3132–3141. [Google Scholar] [CrossRef]
- Simpson, P.J.; Monie, T.P.; Szendröi, A.; Davydova, N.; Tyzack, J.K.; Conte, M.R.; Read, C.M.; Cary, P.D.; Svergun, D.I.; Konarev, P.V.; et al. Structure and RNA interactions of the N-terminal RRM domains of PTB. Structure 2004, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Patton, J.G. Regulation of alternative 3′ splice site selection by constitutive splicing factors. RNA 1995, 1, 234–245. [Google Scholar] [PubMed]
- Singh, R.; Valcárcel, J.; Green, M.R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science 1995, 268, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Garcia-Blanco, M.A. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol. 2001, 21, 3281–3288. [Google Scholar] [CrossRef] [Green Version]
- Chou, M.Y.; Underwood, J.G.; Nikolic, J.; Luu, M.H.; Black, D.L. Multisite RNA binding and release of polypyrimidine tract binding protein during the regulation of c-src neural-specific splicing. Mol. Cell 2000, 5, 949–957. [Google Scholar] [CrossRef]
- Izquierdo, J.M.; Majós, N.; Bonnal, S.; Martínez, C.; Castelo, R.; Guigó, R.; Bilbao, D.; Valcárcel, J. Regulation of fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell 2005, 19, 475–484. [Google Scholar] [CrossRef]
- Cherny, D.; Gooding, C.; Eperon, G.E.; Coelho, M.B.; Bagshaw, C.R.; Smith, C.W.J.; Eperon, I.C. Stoichiometry of a regulatory splicing complex revealed by single-molecule analyses. EMBO J. 2010, 29, 2161–2172. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kohlstaedt, L.A.; Damianov, A.; Rio, D.C.; Black, D.L. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 183–191. [Google Scholar] [CrossRef]
- Sharma, S.; Maris, C.; Allain, F.H.T.; Black, D.L. U1 snRNA Directly Interacts with Polypyrimidine Tract-Binding Protein during Splicing Repression. Mol. Cell 2011, 41, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Polydorides, A.D.; Okano, H.J.; Yang, Y.Y.L.; Stefani, G.; Darnell, R.B. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc. Natl. Acad. Sci. USA 2000, 97, 6350–6355. [Google Scholar] [CrossRef] [Green Version]
- Gromak, N.; Rideau, A.; Southby, J.; Scadden, A.D.J.; Gooding, C.; Hüttelmaier, S.; Singer, R.H.; Smith, C.W.J. The PTB interacting protein raver1 regulates alpha-tropomyosin alternative splicing. EMBO J. 2003, 22, 6356–6364. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, M.G.; Lorenzi, P.; Avesani, F.; Morandi, C. Functional characterization of the ribonucleoprotein, PTB-binding 1/Raver1 promoter region. Gene 2007, 405, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Kleinhenz, B.; Fabienke, M.; Swiniarski, S.; Wittenmayer, N.; Kirsch, J.; Jockusch, B.M.; Arnold, H.H.; Illenberger, S. Raver2, a new member of the hnRNP family. FEBS Lett. 2005, 579, 4254–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanelli, M.G.; Lorenzi, P.; Diani, E.; Filippello, A.; Avesani, F.; Morandi, C. Transcriptional regulation of the human raver2 ribonucleoprotein gene. Gene 2012, 493, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of alternative splicing by histone modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Tsukahara, K.; Kanaoka, Y.; Jinno, S.; Okayama, H. Isolation of a Mammalian Homologue of a Fission Yeast Differentiation Regulator. Mol. Cell. Biol. 1999, 19, 3829–3841. [Google Scholar] [CrossRef] [Green Version]
- Robinson, F.; Jackson, R.J.; Smith, C.W.J. Expression of Human nPTB Is Limited by Extreme Suboptimal Codon Content. PLoS ONE 2008, 3, e1801. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.S.; Inoue, T.; Kulkeaw, K.; Tanaka, Y.; Lai, M.I.; Sugiyama, D. Localized SCF and IGF-1 secretion enhances erythropoiesis in the spleen of murine embryos. Biol. Open 2015, 4, 596–607. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Li, L.; Chen, F.; Chen, Y.; Liu, H.; Li, J.; Bai, J.; Zheng, J. PTBP3-Mediated regulation of zeb1 mRNA stability promotes epithelial–mesenchymal transition in breast cancer. Cancer Res. 2018, 78, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Chen, W.; Shi, H.; Gu, X.; Li, Y.; Qi, Y.; Xu, K.; Zhao, A.; Liu, J. PTBP3 contributes to the metastasis of gastric cancer by mediating CAV1 alternative splicing. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Chen, F.; Yong, H.; Lin, T.; Li, J.; Pan, Y.; Jiang, T.; Li, M.; Chen, Y.; Song, J.; et al. PTBP3 contributes to colorectal cancer growth and metastasis via translational activation of HIF-1α. J. Exp. Clin. Cancer Res. 2019, 38, 301. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Weng, L.; Jia, Y.; Liu, B.; Wu, S.; Xue, L.; Yin, X.; Mao, A.; Wang, Z.; Shang, M. PTBP3 promotes malignancy and hypoxia-induced chemoresistance in pancreatic cancer cells by ATG12 up-regulation. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Fields, P.A.; Ramani, V.; Bonora, G.; Yardimci, G.G.; Reinecke, H.; Pabon, L.; Noble, W.S.; Shendure, J.; Murry, C.E. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Morinaga, A.; Ito, J.; Niimi, T.; Maturana, A.D. RBM20 regulates CaV1.2 surface expression by promoting exon 9* Inclusion of CACNA1C in neonatal rat cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southby, J.; Gooding, C.; Smith, C.W.J. Polypyrimidine Tract Binding Protein Functions as a Repressor To Regulate Alternative Splicing of α-Actinin Mutally Exclusive Exons. Mol. Cell. Biol. 1999, 19, 2699–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollerton, M.C.; Gooding, C.; Robinson, F.; Brown, E.C.; Jackson, R.J.; Smith, C.W.J. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA 2001, 7, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.Z.; Sharma, S.; Zheng, S.; Chawla, G.; Nikolic, J.; Black, D.L. Regulation of the mutually exclusive exons 8a and 8 in the CaV1.2 calcium channel transcript by polypyrimidine tract-binding protein. J. Biol. Chem. 2011, 286, 10007–10016. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Llorian, M.; Cardona, M.; Rongvaux, A.; Moubarak, R.S.; Comella, J.X.; Bassel-Duby, R.; Flavell, R.A.; Olson, E.N.; Smith, C.W.J.; et al. A pathway involving HDAC5, cFLIP and caspases regulates expression of the splicing regulator polypyrimidine tract binding protein in the heart. J. Cell Sci. 2013, 126, 1682–1691. [Google Scholar] [CrossRef] [Green Version]
- Charlet-B, N.; Logan, P.; Singh, G.; Cooper, T.A. Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol. Cell 2002, 9, 649–658. [Google Scholar] [CrossRef]
- Gooding, C.; Roberts, G.C.; Smith, C.W.J. Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated α-tropomyosin exon. RNA 1998, 4, 85–100. [Google Scholar] [CrossRef]
- Mulligan, G.J.; Guo, W.; Wormsley, S.; Helfman, D.M. Polypyrimidine tract binding protein interacts with sequences involved in alternative splicing of beta-tropomyosin pre-mRNA. J. Biol. Chem. 1992, 267, 25480–25487. [Google Scholar] [PubMed]
- LeWinter, M.M.; Granzier, H.L. Titin is a major human disease gene. Circulation 2013, 127, 938–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, C.; Granzier, H. Tuning the molecular giant titin through phosphorylation: Role in health and disease. Trends Cardiovasc. Med. 2013, 23, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Labeit, S.; Kolmerer, B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science 1995, 270, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Linke, W.A.; Hamdani, N. Gigantic business: Titin properties and function through thick and thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Freiburg, A.; Trombitas, K.; Hell, W.; Cazorla, O.; Fougerousse, F.; Centner, T.; Kolmerer, B.; Witt, C.; Beckmann, J.S.; Gregorio, C.C.; et al. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ. Res. 2000, 86, 1114–1121. [Google Scholar] [CrossRef] [Green Version]
- Granzier, H.L.; Labeit, S. The Giant Protein Titin: A Major Player in Myocardial Mechanics, Signaling, and Disease. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Lewinter, M.M. Acute pericarditis. N. Engl. J. Med. 2014, 371, 2410–2416. [Google Scholar] [CrossRef]
- Guo, W.; Zhu, C.; Yin, Z.; Wang, Q.; Sun, M.; Cao, H.; Greaser, M.L. Splicing factor RBM20 regulates transcriptional network of titin associated and calcium handling genes in the heart. Int. J. Biol. Sci. 2018, 14, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Linke, W.A. Plasticity of Cardiac Titin/Connectin in Heart Development. J. Muscle Res. Cell Motil. 2005, 26, 333–342. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.P.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement from the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Pleitner, J.M.; Saupe, K.W.; Greaser, M.L. Pathophysiological defects and transcriptional profiling in the RBM20 -/- rat model. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Guo, W.; Schmitt, B.M.; Greaser, M.L. Comprehensive analysis of titin protein isoform and alternative splicing in normal and mutant rats. J. Cell. Biochem. 2012, 113, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Babicz, K.; Von Frieling-Salewsky, M.; Linke, W.A. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 48, 910–916. [Google Scholar] [CrossRef]
- Zhu, C.; Yin, Z.; Ren, J.; McCormick, R.J.; Ford, S.P.; Guo, W. RBM20 is an essential factor for thyroid hormone-regulated titin isoform transition. J. Mol. Cell Biol. 2015, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Yin, Z.; Tan, B.; Guo, W. Insulin regulates titin pre-mRNA splicing through the PI3K-Akt-mTOR kinase axis in a RBM20-dependent manner. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2363–2371. [Google Scholar] [CrossRef]
- Cai, H.; Zhu, C.; Chen, Z.; Maimaiti, R.; Sun, M.; McCormick, R.J.; Lan, X.; Chen, H.; Guo, W. Angiotensin ii influences pre-mRNA splicing regulation by enhancing RBM20 transcription through activation of the MAPK/ELK1 signaling pathway. Int. J. Mol. Sci. 2019, 20, 5059. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.F.; Reckman, Y.J.; Aufiero, S.; Van Den Hoogenhof, M.M.G.; Van Der Made, I.; Beqqali, A.; Koolbergen, D.R.; Rasmussen, T.B.; Van Der Velden, J.; Creemers, E.E.; et al. RBM20 Regulates Circular RNA Production from the Titin Gene. Circ. Res. 2016, 119, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA Biogenesis competes with Pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Structure and Regulation of Voltage-Gated Ca2+ Channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dai, D.F.; Yuan, C.; Westenbroek, R.E.; Yu, H.; West, N.; De La Iglesia, H.O.; Catterall, W.A. Loss of β-adrenergic-stimulated phosphorylation of CaV1.2 channels on Ser1700 leads to heart failure. Proc. Natl. Acad. Sci. USA 2016, 113, E7976–E7985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Chen, J.; Qin, Y.; Wang, J.; Zhou, L. Mutations in voltage-gated L-type calcium channel: Implications in cardiac arrhythmia. Channels 2018, 12, 201–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, C.B.B.; Heller Brown, J. CaMKIIdelta subtypes: Localization and function. Front. Pharmacol. 2014, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; Van Der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; Van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- George, C.H.; Rogers, S.A.; Bertrand, B.M.A.; Tunwell, R.E.A.; Thomas, N.L.; Steele, D.S.; Cox, E.V.; Pepper, C.; Hazeel, C.J.; Claycomb, W.C.; et al. Alternative splicing of ryanodine receptors modulates cardiomyocyte Ca2+ signaling and susceptibility to apoptosis. Circ. Res. 2007, 100, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Morales, A.; Gonzalez-Quintana, J.; Norton, N.; Siegfried, J.D.; Hofmeyer, M.; Hershberger, R.E. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin. Transl. Sci. 2010, 3, 90–97. [Google Scholar] [CrossRef]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Hear. Rhythm 2012, 9, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Beqqali, A.; Bollen, I.A.E.; Rasmussen, T.B.; Van den Hoogenhof, M.M.; Van Deutekom, H.W.M.; Schafer, S.; Haas, J.; Meder, B.; Sørensen, K.E.; Van Oort, R.J.; et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res. 2016, 112, 452–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Methawasin, M.; Hutchinson, K.R.; Lee, E.J.; Smith, J.E.; Saripalli, C.; Hidalgo, C.G.; Ottenheijm, C.A.C.; Granzier, H. Experimentally increasing titin compliance in a novel mouse model attenuates the frank-starling mechanism but has a beneficial effect on diastole. Circulation 2014, 129, 1924–1936. [Google Scholar] [CrossRef] [PubMed]
- Wyles, S.P.; Li, X.; Hrstka, S.C.; Reyes, S.; Oommen, S.; Beraldi, R.; Edwards, J.; Terzic, A.; Olson, T.M.; Nelson, T.J. Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum. Mol. Genet. 2016, 25, 254–265. [Google Scholar] [CrossRef] [Green Version]
- Streckfuss-Bömeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Özcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Bull, M.; Methawasin, M.; Strom, J.; Nair, P.; Hutchinson, K.; Granzier, H. Alternative splicing of titin restores diastolic function in an HFpEF-like genetic murine model (Ttn ΔIAjxn). Circ. Res. 2016, 119, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinze, F.; Dieterich, C.; Radke, M.H.; Granzier, H.; Gotthardt, M. Reducing RBM20 activity improves diastolic dysfunction and cardiac atrophy. J. Mol. Med. 2016, 94, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Llorian, M.; Gooding, C.; Bellora, N.; Hallegger, M.; Buckroyd, A.; Wang, X.; Rajgor, D.; Kayikci, M.; Feltham, J.; Ule, J.; et al. The alternative splicing program of differentiated smooth muscle cells involves concerted non-productive splicing of post-transcriptional regulators. Nucleic Acids Res. 2016, 44, 8933–8950. [Google Scholar] [CrossRef] [Green Version]
- Llorian, M.; Schwartz, S.; Clark, T.A.; Hollander, D.; Tan, L.Y.; Spellman, R.; Gordon, A.; Schweitzer, A.C.; De La Grange, P.; Ast, G.; et al. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat. Struct. Mol. Biol. 2010, 17, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Spellman, R.; Llorian, M.; Smith, C.W.J. Crossregulation and Functional Redundancy between the Splicing Regulator PTB and Its Paralogs nPTB and ROD1. Mol. Cell 2007, 27, 420–434. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, Y.; Wu, T.; Zhu, T.; Ji, X.; Kwon, Y.S.; Zhang, C.; Yeo, G.; Black, D.L.; Sun, H.; et al. Genome-wide Analysis of PTB-RNA Interactions Reveals a Strategy Used by the General Splicing Repressor to Modulate Exon Inclusion or Skipping. Mol. Cell 2009, 36, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutz, P.L.; Chawla, G.; Stoilov, P.; Black, D.L. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev. 2007, 21, 71–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.C.; Tarn, W.Y. RBM4 down-regulates PTB and antagonizes its activity in muscle cell-specific alternative splicing. J. Cell Biol. 2011, 193, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, D.K.; McLean, M.D.; Dube, S.; Poiesz, B.J. Translational control of tropomyosin expression in vertebrate hearts. Anat. Rec. 2014, 297, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.G.; Mayer, S.A.; Tempst, P.; Nadal-Ginard, B. Characterization and molecular cloning of polypyrimidine tract-binding protein: A component of a complex necessary for pre-mRNA splicing. Genes Dev. 1991, 5, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorian, M.; Smith, C.W.J. Decoding muscle alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 380–387. [Google Scholar] [CrossRef]
- McConnell, M.; Tal Grinspan, L.; Williams, M.R.; Lynn, M.L.; Schwartz, B.A.; Fass, O.Z.; Schwartz, S.D.; Tardiff, J.C. Clinically Divergent Mutation Effects on the Structure and Function of the Human Cardiac Tropomyosin Overlap. Biochemistry 2017, 56, 3403–3413. [Google Scholar] [CrossRef]
- Godt, R.E.; Fogaça, R.T.; Silva, I.K.; Nosek, T.M. Contraction of developing avian heart muscle. Comp. Biochem. Physiol. Comp. Physiol. 1993, 105, 213–218. [Google Scholar] [CrossRef]
- Cooper, T.A.; Ordahl, C.P. A single cardiac troponin T gene generates embryonic and adult isoforms via developmentally regulated alternate splicing. J. Biol. Chem. 1985, 260, 11140–11148. [Google Scholar]
- Ladd, A.N.; Stenberg, M.G.; Swanson, M.S.; Cooper, T.A. Dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Dev. Dyn. 2005, 233, 783–793. [Google Scholar] [CrossRef]
- Waites, G.T.; Graham, I.R.; Jackson, P.; Millake, D.B.; Patel, B.; Blanchard, A.D.; Weller, P.A.; Eperon, I.C.; Critchley, D.R. Mutually exclusive splicing of calcium-binding domain exons in chick alpha-actinin. J. Biol. Chem. 1992, 267, 6263–6271. [Google Scholar] [PubMed]
- Matlin, A.J.; Southby, J.; Gooding, C.; Smith, C.W.J. Repression of α-actinin SM exon splicing by assisted binding of PTB to the polypyrimidine tract. RNA 2007, 13, 1214–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Kim, J.O.; Oh, J.G.; Hong, S.E.; Kim, D.H. Pressure-overload cardiac hypertrophy is associated with distinct alternative splicing due to altered expression of splicing factors. Mol. Cells 2014, 37, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan-o, M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian formin Fhod3 regulates actin assembly and sarcomere organization in striated muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan-O, M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The actin-organizing formin protein Fhod3 is required for postnatal development and functional maintenance of the adult heart in mice. J. Biol. Chem. 2018, 293, 148–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten, E.C.; Hebl, V.B.; Wolf, M.J.; Greytak, S.R.; Orr, N.M.; Draper, I.; Calvino, J.E.; Kapur, N.K.; Maron, M.S.; Kullo, I.J.; et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 2013, 77, 2990–2996. [Google Scholar] [CrossRef] [Green Version]
- Beraldi, R.; Li, X.; Fernandez, A.M.; Reyes, S.; Secreto, F.; Terzic, A.; Olson, T.M.; Nelson, T.J. Rbm20-deficient cardiogenesis reveals early disruption of RNA processing and sarcomere remodeling establishing a developmental etiology for dilated cardiomyopathy. Hum. Mol. Genet. 2014, 23, 3779–3791. [Google Scholar] [CrossRef] [Green Version]
- Lara-Pezzi, E.; Gómez-Salinero, J.; Gatto, A.; García-Pavía, P. The alternative heart: Impact of alternative splicing in heart disease. J. Cardiovasc. Transl. Res. 2013, 6, 945–955. [Google Scholar] [CrossRef]
- Kong, S.W.; Hu, Y.W.; Ho, J.W.K.; Ikeda, S.; Polster, S.; John, R.; Hall, J.L.; Bisping, E.; Pieske, B.; Dos Remedios, C.G.; et al. Heart Failure-Associated Changes in RNA Splicing of Sarcomere Genes. Circ. Cardiovasc. Genet. 2010, 3, 138–146. [Google Scholar] [CrossRef]
- Giudice, J.; Xia, Z.; Wang, E.T.; Scavuzzo, M.A.; Ward, A.J.; Kalsotra, A.; Wang, W.; Wehrens, X.H.T.; Burge, C.B.; Li, W.; et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 2014, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ito, J.; Iijima, M.; Yoshimoto, N.; Niimi, T.; Kuroda, S.; Maturana, A.D. RBM20 and RBM24 cooperatively promote the expression of short enh splice variants. FEBS Lett. 2016, 590, 2262–2274. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hung, L.H.; Licht, T.; Kostin, S.; Looso, M.; Khrameeva, E.; Bindereif, A.; Schneider, A.; Braun, T. RBM24 Is a major regulator of muscle-specific alternative splicing. Dev. Cell 2014, 31, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhang, Y.; Xu, E.; Mohibi, S.; De Anda, D.M.; Jiang, Y.; Zhang, J.; Chen, X. Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ. 2018, 25, 1118–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, K.L.; Tan, K.T.; Wei, Y.Y.; Ng, C.P.; Colman, A.; Korzh, V.; Xu, X.Q. RNA-binding protein RBM24 is required for sarcomere assembly and heart contractility. Cardiovasc. Res. 2012, 94, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kong, X.; Zhang, M.; Yang, X.; Xu, X. RNA binding protein 24 deletion disrupts global alternative splicing and causes dilated cardiomyopathy. Protein Cell 2019, 10, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Gaertner, A.; Brodehl, A.; Milting, H. Screening for mutations in human cardiomyopathy- is RBM24 a new but rare disease gene? Protein Cell 2019, 10, 393–394. [Google Scholar] [CrossRef] [Green Version]
- Maturana, A.D.; Nakagawa, N.; Yoshimoto, N.; Tatematsu, K.; Hoshijima, M.; Tanizawa, K.; Kuroda, S. LIM domains regulate protein kinase C activity: A novel molecular function. Cell. Signal. 2011, 23, 928–934. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Gene Symbola | Gene Name | Regulated Exons | Species | References |
|---|---|---|---|---|
| RBM20-regulated genes | ||||
| CACNA1C | Calcium Voltage-Gated Channel Subunit α1 C | 9 | Human ESC RBM20 KO CMs | [88] |
| 9* | Rat cardiomyocytes | [89] | ||
| CAMK2D | Calcium/calmodulin dependent protein kinase II delta | 14 | Human cardiac RBM20 (S635A) tissue, Rbm20-/- rats, Human ESC RBM20 KO CMs | [33] |
| CAMKIIG | Calcium/calmodulin dependent protein kinase II gamma | 12–15 | Human cardiac RBM20 (S635A) tissue, Rbm20-/- rats | [33] |
| FHOD3 | Formin homology 2 domain containing 3 | 12–14 | HeLa cells | [32] |
| LDB3 | LIM domain binding 3 | 4, 5/6 | Human cardiac RBM20 (S635A) tissue, Rbm20-/- rats | [33] |
| LMO7 | LIM domain only protein 7 | 9, 10 | Rbm20-/- rats | [38] |
| MLIP | Muscular-enriched A type laminin-interacting protein | 9, 10 | Rbm20-/- rats | [38] |
| PDLIM3 | PDZ and LIM domain 3 | 4–6 | Rbm20-/- rats | [38] |
| RTN4 | Reticulon 4 | 3, 4 | Rbm20-/- rats | [38] |
| RYR2 | Ryanodine receptor 2 | 24 bp exon | Rbm20-/- rats | [38] |
| SH3KBP1 | SH3 domain containing kinase binding protein 1 | 6–7 | Human cardiac RBM20 (S635A) tissue, Rbm20-/- rats | [33] |
| SORBS1 | Sorbin and SH3 domain containing 1 | 2 | Human cardiac RBM20 (S635A) tissue, Rbm20-/- rats | [33] |
| TRDN | Triadin | 8 | Human cardiac RBM20 (S635A) tissue, Rbm20-/- rats | [33] |
| TTN | Titin | PEVK exons | Human cardiac RBM20 (S635A) tissue, Rbm20-/- rats, Human ESC RBM20 KO CMs | [33,88] |
| PTBP1-regulated genes | ||||
| ACTN1 | α-actinin | NM/SM | Non-smooth muscle cells, PAC1 smooth muscle cells | [90,91] |
| CACNA1C | Calcium Voltage-Gated Channel Subunit α1 C | 8/8a | Neuro-2a cells | [92] |
| FHOD3 | Formin homology 2 domain containing 3 | 12–14 | HeLa cells | [32] |
| MEF2 | Myocyte Enhancer Factor 2 | 3 | Rat cardiomyocytes | [93] |
| TNNT2 | Troponin T type 2 | 5 | Primary embryonic skeletal muscle cultures | [94] |
| TPM1 | Tropomyosin 1 | 3 | PAC1 smooth muscle cells | [91,95] |
| 9 | Rat cardiomyocytes | [93] | ||
| TPM2 | Tropomyosin 2 | 7 | HeLa cells, rat cardiomyocytes | [93,96] |
| TTN | Titin | 242 | HEK293 cells | [31] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fochi, S.; Lorenzi, P.; Galasso, M.; Stefani, C.; Trabetti, E.; Zipeto, D.; Romanelli, M.G. The Emerging Role of the RBM20 and PTBP1 Ribonucleoproteins in Heart Development and Cardiovascular Diseases. Genes 2020, 11, 402. https://doi.org/10.3390/genes11040402
Fochi S, Lorenzi P, Galasso M, Stefani C, Trabetti E, Zipeto D, Romanelli MG. The Emerging Role of the RBM20 and PTBP1 Ribonucleoproteins in Heart Development and Cardiovascular Diseases. Genes. 2020; 11(4):402. https://doi.org/10.3390/genes11040402
Chicago/Turabian StyleFochi, Stefania, Pamela Lorenzi, Marilisa Galasso, Chiara Stefani, Elisabetta Trabetti, Donato Zipeto, and Maria Grazia Romanelli. 2020. "The Emerging Role of the RBM20 and PTBP1 Ribonucleoproteins in Heart Development and Cardiovascular Diseases" Genes 11, no. 4: 402. https://doi.org/10.3390/genes11040402