Metabolomics: A Tool to Understand the Impact of Genetic Mutations in Amyotrophic Lateral Sclerosis

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Overview of ALS Genetics
2.1. C9ORF72
2.2. SOD1
2.3. TARDBP
2.4. FUS

{kind=link}
| Gene | Function of Coded Protein | fALS/sALS Cases (%) | Alteration in Metabolome | Model |
|---|---|---|---|---|
| C9ORF72 | Autophagy–lysosome pathway | 34/5 | ↓HDL | FTLD [49] |
| SOD1 | Antioxidant | 15-20/2 | ↓ aminoacids ↓ aminoacids; ↑ glycolysis ↓ glutamate ↑ putrescine and spermidine; ↓ hydroxyproline ↑ creatinine | ALS patients [50] NSC-34 cells [51] Motor neuron/ astrocytes cultures [52] mice [53] ALS patients [53] |
| TARDBP | RNA metabolism | 3/1.5 | ↓ carnitine and beta-hydroxybutyrate ↑ phosphoenolpyruvate and pyruvate ↑ fatty acids | Drosophila [54] Drosophila [55] HEK293T cells [56] |
| FUS | RNA metabolism | 2.4/0.16 [57] 3.8 [58] 4.1 [42] | none | iPSC-derived motor neurons [59] |
| SNP rs1985243 | not described | - | ↑ gamma-glutamylphenylalanine | ALS patients [60] |
3. Metabolomics and Genetic-Linked ALS: A Way into the Targeted Treatment
3.1. C9ORF72
3.2. SOD1
3.3. TARDBP
3.4. FUS
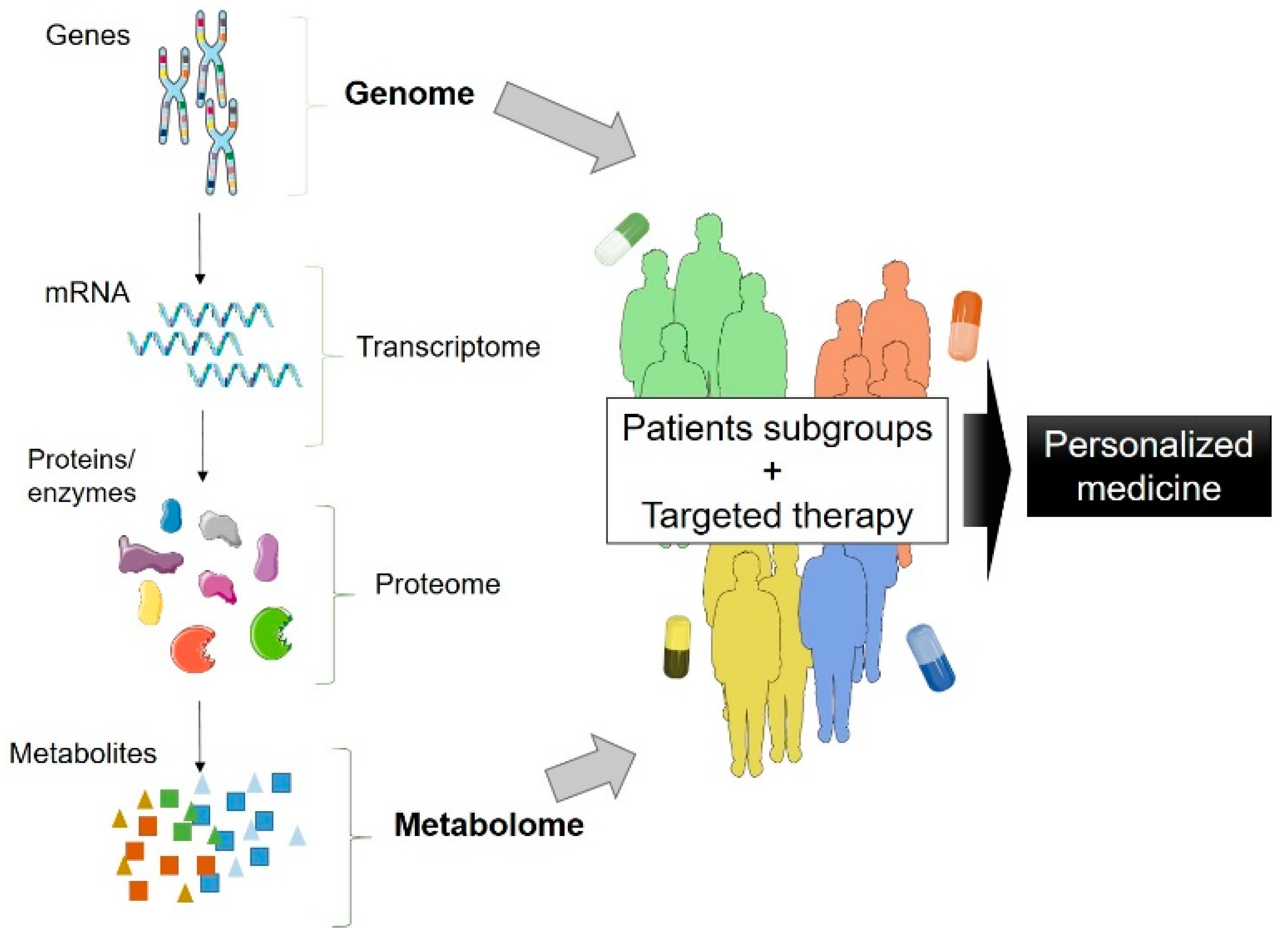
4. Combination of Omics and Patient-Derived iPSC: The Future of ALS Research
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chio, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsultan, A.A.; Waller, R.; Heath, P.R.; Kirby, J. The genetics of amyotrophic lateral sclerosis: Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 49–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative genes in amyotrophic lateral sclerosis and protein degradation pathways: A link to neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Ferri, A.; Coccurello, R. What is "Hyper" in the ALS hypermetabolism? Mediat. Inflamm. 2017, 2017, 7821672. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Jesus, P.; Fayemendy, P.; Nicol, M.; Lautrette, G.; Sourisseau, H.; Preux, P.M.; Desport, J.C.; Marin, B.; Couratier, P. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2018, 25, 97–104. [Google Scholar] [CrossRef]
- Brito, M.D.; da Silva, G.F.G.; Tilieri, E.M.; Araujo, B.G.; Calio, M.L.; Rosenstock, T.R. Metabolic alteration and amyotrophic lateral sclerosis outcome: A systematic review. Front. Neurol. 2019, 10, 1205. [Google Scholar] [CrossRef] [Green Version]
- Boylan, K.B.; Glass, J.D.; Crook, J.E.; Yang, C.; Thomas, C.S.; Desaro, P.; Johnston, A.; Overstreet, K.; Kelly, C.; Polak, M.; et al. Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and CSF as a potential prognostic biomarker in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 467–472. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, mechanisms, and therapeutics: Where are we now? Front. Mol. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Nassif, M.; Woehlbier, U.; Manque, P.A. The enigmatic role of C9ORF72 in autophagy. Front. Mol. Neurosci. 2017, 11, 442. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J. C9orf72-dependent lysosomal functions regulate epigenetic control of autophagy and lipid metabolism. Autophagy 2019, 15, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.; Vieira de Sa, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Burberry, A.; Suzuki, N.; Wang, J.Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016, 8, 347ra393. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 Is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.G.; Bogdanik, L.; Yanez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudria-Lopez, E.; Koppers, M.; de Wit, M.; van der Meer, C.; Westeneng, H.J.; Zundel, C.A.; Youssef, S.A.; Harkema, L.; de Bruin, A.; Veldink, J.H.; et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016, 132, 145–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- May, S.; Hornburg, D.; Schludi, M.H.; Arzberger, T.; Rentzsch, K.; Schwenk, B.M.; Grasser, F.A.; Mori, K.; Kremmer, E.; Banzhaf-Strathmann, J.; et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014, 128, 485–503. [Google Scholar] [CrossRef] [Green Version]
- Mizielinska, S.; Gronke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 2016, 167, 774–788 e717. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 2016, 167, 789–802 e712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Z.; Liu, L.; Tao, Z.; Wang, R.; Ren, H.; Sun, H.; Lin, Z.; Zhang, Z.; Mu, C.; Zhou, J.; et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun. 2019, 10, 2906. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M.; Graslund, A.; Reichard, P. The function of superoxide dismutase during the enzymatic formation of the free radical of ribonucleotide reductase. J. Biol. Chem. 1987, 262, 12332–12336. [Google Scholar] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Felbecker, A.; Camu, W.; Valdmanis, P.N.; Sperfeld, A.D.; Waibel, S.; Steinbach, P.; Rouleau, G.A.; Ludolph, A.C.; Andersen, P.M. Four familial ALS pedigrees discordant for two SOD1 mutations: Are all SOD1 mutations pathogenic? J. Neurol. Neurosurg. Psychiatry 2010, 81, 572–577. [Google Scholar] [CrossRef]
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. NeuroDegener. 2015, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. BioChem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.Y.; Cui, L.Y.; Sun, Q.; Li, X.G.; Liu, M.S.; Xu, Y.; Zhou, Y.; Yang, X.Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. NeuroBiol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Hubers, A.; Just, W.; Rosenbohm, A.; Muller, K.; Marroquin, N.; Goebel, I.; Hogel, J.; Thiele, H.; Altmuller, J.; Nurnberg, P.; et al. De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. NeuroBiol. Aging 2015, 36, 3117.e1–3117.e6. [Google Scholar] [CrossRef] [PubMed]
- Gromicho, M.; Oliveira Santos, M.; Pinto, A.; Pronto-Laborinho, A.; De Carvalho, M. Young-onset rapidly progressive ALS associated with heterozygous FUS mutation. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110 Pt 15, 1741–1750. [Google Scholar]
- Niu, C.; Zhang, J.; Gao, F.; Yang, L.; Jia, M.; Zhu, H.; Gong, W. FUS-NLS/Transportin 1 complex structure provides insights into the nuclear targeting mechanism of FUS and the implications in ALS. PLoS ONE 2012, 7, e47056. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobagyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef]
- Jaaskelainen, O.; Solje, E.; Hall, A.; Katisko, K.; Korhonen, V.; Tiainen, M.; Kangas, A.J.; Helisalmi, S.; Pikkarainen, M.; Koivisto, A.; et al. Low serum high-density lipoprotein cholesterol levels associate with the C9orf72 repeat expansion in frontotemporal lobar degeneration patients. J. Alzheimers Dis. 2019, 72, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Wuolikainen, A.; Andersen, P.M.; Moritz, T.; Marklund, S.L.; Antti, H. ALS patients with mutations in the SOD1 gene have an unique metabolomic profile in the cerebrospinal fluid compared with ALS patients without mutations. Mol. Genet. Metab. 2012, 105, 472–478. [Google Scholar] [CrossRef]
- Valbuena, G.N.; Rizzardini, M.; Cimini, S.; Siskos, A.P.; Bendotti, C.; Cantoni, L.; Keun, H.C. Metabolomic analysis reveals increased aerobic glycolysis and amino acid deficit in a cellular model of amyotrophic lateral sclerosis. Mol. Neurobiol. 2016, 53, 2222–2240. [Google Scholar] [CrossRef] [Green Version]
- Valbuena, G.N.; Tortarolo, M.; Bendotti, C.; Cantoni, L.; Keun, H.C. Altered metabolic profiles associate with toxicity in SOD1G93A astrocyte-neuron co-cultures. Sci. Rep. 2017, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Patin, F.; Corcia, P.; Vourc’h, P.; Nadal-Desbarats, L.; Baranek, T.; Goossens, J.F.; Marouillat, S.; Dessein, A.F.; Descat, A.; Madji Hounoum, B.; et al. Omics to explore amyotrophic lateral sclerosis evolution: The central role of arginine and proline metabolism. Mol. Neurobiol. 2017, 54, 5361–5374. [Google Scholar] [CrossRef]
- Manzo, E.; O’Conner, A.G.; Barrows, J.M.; Shreiner, D.D.; Birchak, G.J.; Zarnescu, D.C. Medium-chain fatty acids, beta-hydroxybutyric acid and genetic modulation of the carnitine shuttle are protective in a drosophila model of ALS based on TDP-43. Front. Mol. NeuroSci. 2018, 11, 182. [Google Scholar] [CrossRef]
- Manzo, E.; Lorenzini, I.; Barrameda, D.; O’Conner, A.G.; Barrows, J.M.; Starr, A.; Kovalik, T.; Rabichow, B.E.; Lehmkuhl, E.M.; Shreiner, D.D.; et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife 2019, 8. [Google Scholar] [CrossRef]
- Lanznaster, D.; Bourgeais, J.; Bruno, C.; Hergesheimer, R.C.; Thepault, R.A.; Vourc’h, P.; Corcia, P.; Andres, C.R.; Herault, O.; Blasco, H. TDP-43-mediated toxicity in HEK293T Cells: A fast and reproducible protocol to be employed in the search of new therapeutic options against amyotrophic lateral sclerosis. Cells 2019, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Drepper, C.; Herrmann, T.; Wessig, C.; Beck, M.; Sendtner, M. C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. NeuroBiol. Aging 2011, 32, 548.e1–548.e4. [Google Scholar] [CrossRef]
- Chio, A.; Restagno, G.; Brunetti, M.; Ossola, I.; Calvo, A.; Mora, G.; Sabatelli, M.; Monsurro, M.R.; Battistini, S.; Mandrioli, J.; et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. NeuroBiol. Aging 2009, 30, 1272–1275. [Google Scholar] [CrossRef]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; et al. Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lv, X.; Du, H.; Wu, D.; Wang, M. Causal effects of serum metabolites on amyotrophic lateral sclerosis: A Mendelian randomization study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109771. [Google Scholar] [CrossRef]
- Menni, C.; Zierer, J.; Valdes, A.M.; Spector, T.D. Mixing omics: Combining genetics and metabolomics to study rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, T.; Ji, Y.J.; Johnson, K.; Liu, H.; Johnson, K.; Bailey, S.; Suk, Y.; Lu, Y.N.; Liu, M.; et al. A C9orf72-CARM1 axis regulates lipid metabolism under glucose starvation-induced nutrient stress. Genes Dev. 2018, 32, 1380–1397. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; D’Onofrio, G.; Sancarlo, D.; Greco, A.; Yu, Z. Potential fluid biomarkers for pathological brain changes in Alzheimer’s disease: Implication for the screening of cognitive frailty. Mol. Med. Rep. 2016, 14, 3184–3198. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Yang, J.S.; Lee, J.C.; Lee, J.Y.; Lee, J.Y.; Kim, E.; Moon, M.H. Lipidomic alterations in lipoproteins of patients with mild cognitive impairment and Alzheimer’s disease by asymmetrical flow field-flow fractionation and nanoflow ultrahigh performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1568, 91–100. [Google Scholar] [CrossRef]
- Costa, A.C.; Joaquim, H.P.G.; Forlenza, O.; Talib, L.L.; Gattaz, W.F. Plasma lipids metabolism in mild cognitive impairment and Alzheimer’s disease. World J. Biol. Psychiatry 2019, 20, 190–196. [Google Scholar] [CrossRef]
- Sarrafpour, S.; Ormseth, C.; Chiang, A.; Arakaki, X.; Harrington, M.; Fonteh, A. Lipid metabolism in late-onset Alzheimer’s disease differs from patients presenting with other dementia phenotypes. Int. J. Environ. Res. Public Health 2019, 16, 1995. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M.; Forsgren, L.; Binzer, M.; Nilsson, P.; Ala-Hurula, V.; Keranen, M.L.; Bergmark, L.; Saarinen, A.; Haltia, T.; Tarvainen, I.; et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain 1996, 119 Pt 4, 1153–1172. [Google Scholar] [CrossRef] [Green Version]
- Veyrat-Durebex, C.; Corcia, P.; Piver, E.; Devos, D.; Dangoumau, A.; Gouel, F.; Vourc’h, P.; Emond, P.; Laumonnier, F.; Nadal-Desbarats, L.; et al. Disruption of TCA cycle and glutamate metabolism identified by metabolomics in an in vitro model of amyotrophic lateral sclerosis. Mol. Neurobiol. 2016, 53, 6910–6924. [Google Scholar] [CrossRef]
- Madji Hounoum, B.; Mavel, S.; Coque, E.; Patin, F.; Vourc’h, P.; Marouillat, S.; Nadal-Desbarats, L.; Emond, P.; Corcia, P.; Andres, C.R.; et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia 2017, 65, 592–605. [Google Scholar] [CrossRef]
- Patin, F.; Baranek, T.; Vourc’h, P.; Nadal-Desbarats, L.; Goossens, J.F.; Marouillat, S.; Dessein, A.F.; Descat, A.; Hounoum, B.M.; Bruno, C.; et al. combined metabolomics and transcriptomics approaches to assess the IL-6 blockade as a therapeutic of ALS: Deleterious alteration of lipid metabolism. Neurotherapeutics 2016, 13, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.P. Sphingolipid metabolism is dysregulated at transcriptomic and metabolic levels in the spinal cord of an animal model of amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2017, 10, 433. [Google Scholar] [CrossRef]
- Valbuena, G.N.; Cantoni, L.; Tortarolo, M.; Bendotti, C.; Keun, H.C. spinal cord metabolic signatures in models of fast- and slow-progressing SOD1G93A amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 13, 1276. [Google Scholar] [CrossRef]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P.; et al. Lipidomics reveals cerebrospinal-fluid signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef]
- Gonzalez De Aguilar, J.L. Lipid biomarkers for amyotrophic lateral sclerosis. Front. Neurol. 2019, 10, 284. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Dedic, S.I.; Stevic, Z.; Dedic, V.; Stojanovic, V.R.; Milicev, M.; Lavrnic, D. Is hyperlipidemia correlated with longer survival in patients with amyotrophic lateral sclerosis? Neurol. Res. 2012, 34, 576–580. [Google Scholar] [CrossRef]
- Ikeda, K.; Hirayama, T.; Takazawa, T.; Kawabe, K.; Iwasaki, Y. Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: A cross-sectional study. Intern. Med. 2012, 51, 1501–1508. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, M.K.; Lee, E.; Bradburn, M.; McDermott, C.J.; Shaw, P.J. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: Insights from the olesoxime clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 478–484. [Google Scholar] [CrossRef]
- Delaye, J.B.; Patin, F.; Piver, E.; Bruno, C.; Vasse, M.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Blasco, H. Low IDL-B and high LDL-1 subfraction levels in serum of ALS patients. J. Neurol. Sci. 2017, 380, 124–127. [Google Scholar] [CrossRef]
- Beghi, E.; Pupillo, E.; Bonito, V.; Buzzi, P.; Caponnetto, C.; Chio, A.; Corbo, M.; Giannini, F.; Inghilleri, M.; Bella, V.L.; et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for ALS. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 397–405. [Google Scholar] [CrossRef]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; Hrabe de Angelis, M.; Kronenberg, F.; Meitinger, T.; Mewes, H.W.; Wichmann, H.E.; Weinberger, K.M.; Adamski, J.; et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef] [Green Version]
- Suhre, K.; Shin, S.Y.; Petersen, A.K.; Mohney, R.P.; Meredith, D.; Wagele, B.; Altmaier, E.; CardioGram; Deloukas, P.; Erdmann, J.; et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011, 477, 54–60. [Google Scholar] [CrossRef]
- Kettunen, J.; Tukiainen, T.; Sarin, A.P.; Ortega-Alonso, A.; Tikkanen, E.; Lyytikainen, L.P.; Kangas, A.J.; Soininen, P.; Wurtz, P.; Silander, K.; et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat. Genet. 2012, 44, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef]
- Kumar, D.; Anand, T.; Kues, W.A. Clinical potential of human-induced pluripotent stem cells: Perspectives of induced pluripotent stem cells. Cell Biol. Toxicol. 2017, 33, 99–112. [Google Scholar] [CrossRef]
- Gaignerie, A.; Lefort, N.; Rousselle, M.; Forest-Choquet, V.; Flippe, L.; Francois-Campion, V.; Girardeau, A.; Caillaud, A.; Chariau, C.; Francheteau, Q.; et al. Urine-derived cells provide a readily accessible cell type for feeder-free mRNA reprogramming. Sci. Rep. 2018, 8, 14363. [Google Scholar] [CrossRef] [Green Version]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell NeuroSci. 2013, 56, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Bohl, D.; Pochet, R.; Mitrecic, D.; Nicaise, C. Modelling and treating amyotrophic lateral sclerosis through induced-pluripotent stem cells technology. Curr. Stem Cell Res. Ther. 2016, 11, 301–312. [Google Scholar] [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Sun, X.; Song, J.; Huang, H.; Chen, H.; Qian, K. Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells. Stem Cell Res. Ther. 2018, 9, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 2019, 142, 3771–3790. [Google Scholar] [CrossRef] [PubMed]
- Lawton, K.A.; Brown, M.V.; Alexander, D.; Li, Z.; Wulff, J.E.; Lawson, R.; Jaffa, M.; Milburn, M.V.; Ryals, J.A.; Bowser, R.; et al. Plasma metabolomic biomarker panel to distinguish patients with amyotrophic lateral sclerosis from disease mimics. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 362–370. [Google Scholar] [CrossRef]
- Kori, M.; Aydin, B.; Unal, S.; Arga, K.Y.; Kazan, D. Metabolic biomarkers and neurodegeneration: A pathway enrichment analysis of Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Omics 2016, 20, 645–661. [Google Scholar] [CrossRef]
- Huang, X.; Ng, S.Y.; Chia, N.S.; Acharyya, S.; Setiawan, F.; Lu, Z.H.; Ng, E.; Tay, K.Y.; Au, W.L.; Tan, E.K.; et al. Serum uric acid level and its association with motor subtypes and non-motor symptoms in early Parkinson’s disease: PALS study. Park. Relat. Disord. 2018, 55, 50–54. [Google Scholar] [CrossRef]
- Bjornevik, K.; Zhang, Z.; O’Reilly, E.J.; Berry, J.D.; Clish, C.B.; Deik, A.; Jeanfavre, S.; Kato, I.; Kelly, R.S.; Kolonel, L.N.; et al. Prediagnostic plasma metabolomics and the risk of amyotrophic lateral sclerosis. Neurology 2019, 92, e2089–e2100. [Google Scholar] [CrossRef] [Green Version]
- Corey-Bloom, J.; Haque, A.; Aboufadel, S.; Snell, C.; Fischer, R.S.; Granger, S.W.; Granger, D.A.; Thomas, E.A. Uric acid as a potential peripheral biomarker for disease features in Huntington’s patients. Front. Mol. Neurosci. 2020, 14, 73. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhang, Q.; Ke, Y.; Hao, J.; Lu, L.; Lu, N.; Chen, X. Serum uric acid levels in patients with amyotrophic lateral sclerosis: A meta-analysis. Sci. Rep. 2018, 8, 1100. [Google Scholar] [CrossRef]
- Bakshi, R.; Xu, Y.; Mueller, K.A.; Chen, X.; Granucci, E.; Paganoni, S.; Sadri-Vakili, G.; Schwarzschild, M.A. Urate mitigates oxidative stress and motor neuron toxicity of astrocytes derived from ALS-linked SOD1G93A mutant mice. Mol. Cell. Neurosci. 2018, 92, 12–16. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Liang, W.; Wang, T.; Wang, S.; Wang, X.; Wang, Y.; Jiang, H.; Feng, H. Neuroprotection by urate on the mutant hSOD1-related cellular and Drosophila models of amyotrophic lateral sclerosis: Implication for GSH synthesis via activating Akt/GSK3beta/Nrf2/GCLC pathways. Brain Res. Bull. 2019, 146, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Keizman, D.; Ish-Shalom, M.; Berliner, S.; Maimon, N.; Vered, Y.; Artamonov, I.; Tsehori, J.; Nefussy, B.; Drory, V.E. Low uric acid levels in serum of patients with ALS: Further evidence for oxidative stress? J. Neurol. Sci. 2009, 285, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Yamamoto, Y.; Miyazaki, Y.; Yoshino, H. Increased oxidative stress in patients with amyotrophic lateral sclerosis and the effect of edaravone administration. Redox Rep. 2016, 21, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in oxidative stress markers in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Oxidative Med. Cell. Longev. 2019, 2019, 1712323. [Google Scholar] [CrossRef]
- Nicholson, K.; Chan, J.; Macklin, E.A.; Levine-Weinberg, M.; Breen, C.; Bakshi, R.; Grasso, D.L.; Wills, A.M.; Jahandideh, S.; Taylor, A.A.; et al. Pilot trial of inosine to elevate urate levels in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 1522–1533. [Google Scholar] [CrossRef] [Green Version]
- Lanznaster, D.; de Assis, D.R.; Corcia, P.; Pradat, P.F.; Blasco, H. Metabolomics biomarkers: A strategy toward therapeutics improvement in ALS. Front. Neurol. 2018, 9, 1126. [Google Scholar] [CrossRef] [Green Version]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62 e48. [Google Scholar] [CrossRef]
- Laiko, V.V.; Baldwin, M.A.; Burlingame, A.L. Atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 2000, 72, 652–657. [Google Scholar] [CrossRef]
- Emara, S.; Amer, S.; Ali, A.; Abouleila, Y.; Oga, A.; Masujima, T. Single-cell metabolomics. Adv. Exp. Med. Biol. 2017, 965, 323–343. [Google Scholar] [CrossRef]
- Chappell, L.; Russell, A.J.C.; Voet, T. Single-cell (Multi)omics technologies. Ann. Rev. Genom. Hum. Genet. 2018, 19, 15–41. [Google Scholar] [CrossRef]
- Qi, M.; Philip, M.C.; Yang, N.; Sweedler, J.V. Single cell neurometabolomics. ACS Chem. NeuroSci. 2018, 9, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Germeys, C.; Vandoorne, T.; Bercier, V.; Van Den Bosch, L. Existing and emerging metabolomic tools for ALS research. Genes 2019, 10, 1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, B. Single-cell metabolomics by mass spectrometry. Methods Mol. Biol. 2020, 2064, 1–8. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanznaster, D.; Veyrat-Durebex, C.; Vourc’h, P.; Andres, C.R.; Blasco, H.; Corcia, P. Metabolomics: A Tool to Understand the Impact of Genetic Mutations in Amyotrophic Lateral Sclerosis. Genes 2020, 11, 537. https://doi.org/10.3390/genes11050537
Lanznaster D, Veyrat-Durebex C, Vourc’h P, Andres CR, Blasco H, Corcia P. Metabolomics: A Tool to Understand the Impact of Genetic Mutations in Amyotrophic Lateral Sclerosis. Genes. 2020; 11(5):537. https://doi.org/10.3390/genes11050537
Chicago/Turabian StyleLanznaster, Débora, Charlotte Veyrat-Durebex, Patrick Vourc’h, Christian R. Andres, Hélène Blasco, and Philippe Corcia. 2020. "Metabolomics: A Tool to Understand the Impact of Genetic Mutations in Amyotrophic Lateral Sclerosis" Genes 11, no. 5: 537. https://doi.org/10.3390/genes11050537
APA StyleLanznaster, D., Veyrat-Durebex, C., Vourc’h, P., Andres, C. R., Blasco, H., & Corcia, P. (2020). Metabolomics: A Tool to Understand the Impact of Genetic Mutations in Amyotrophic Lateral Sclerosis. Genes, 11(5), 537. https://doi.org/10.3390/genes11050537