Exome-Wide Analysis of the DiscovEHR Cohort Reveals Novel Candidate Pharmacogenomic Variants for Clinical Pharmacogenomics

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Variant Annotation
2.3. Analysis of Annotated Variants
3. Results
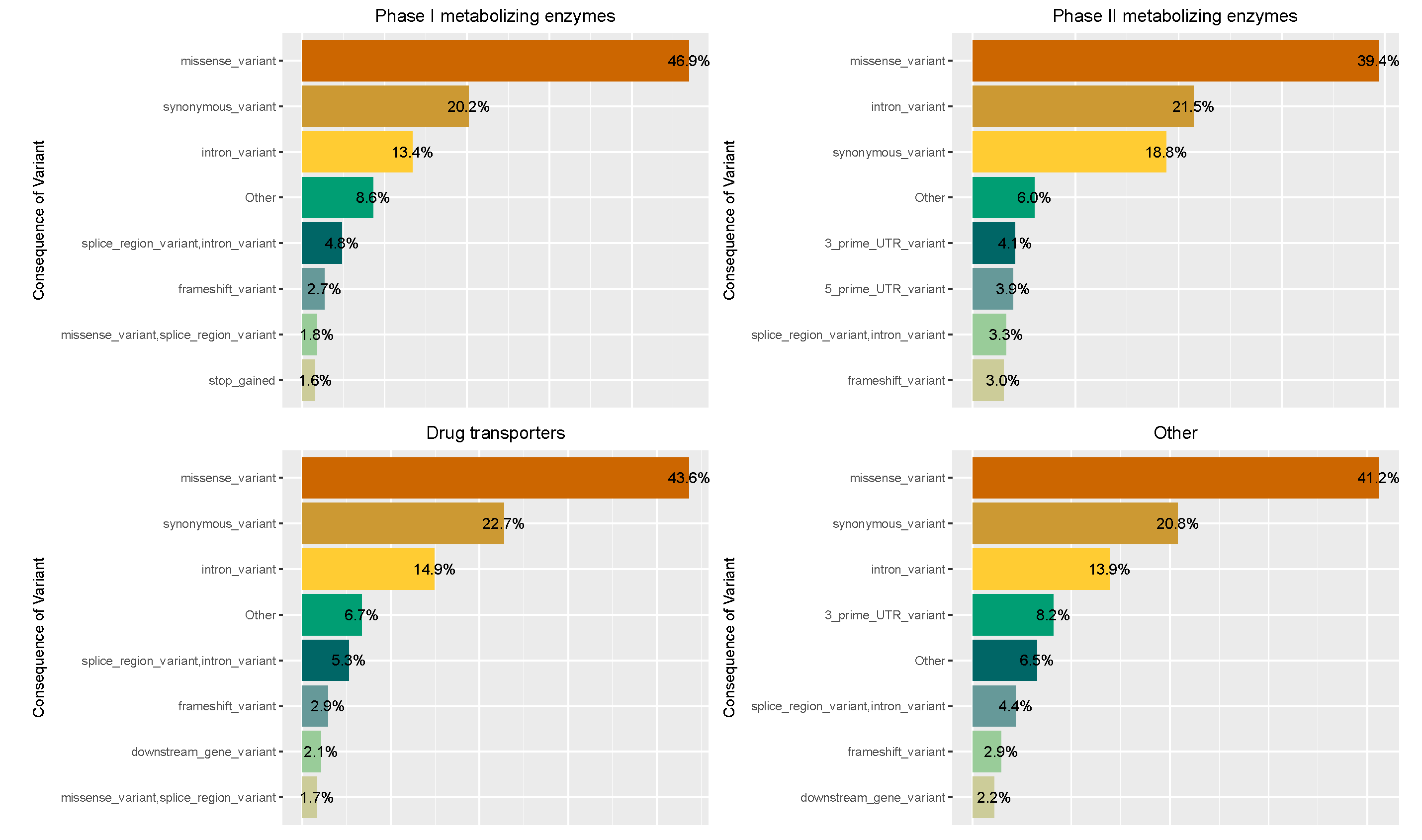
3.1. Composition of Protein-Coding PGx-Related Variation in 50,726 Exomes
3.2. Distribution of the Frequencies of the Identified PGx Variants within Different gnomAD Populations
3.3. Assessing the Protein Damaging Effect of Variants in the 231 DMET Genes within the DiscovEHR Cohort
3.4. Assessing the Pharmacogenomics Clinical Relevance of the Identified PGx Variants
3.5. Assessing LoF PGx Variants within 50,726 Exomes
4. Discussion
4.1. Rare PGx Variation within the DiscovEHR Cohort
4.2. Identifying Ultra-Rare Damaging PGx Variants within the DiscovEHR Cohort
4.3. Towards Clinical Pharmacogenomics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carey, D.; Fetterolf, S.N.; Davis, F.D.; Faucett, W.; Kirchner, H.L.; Mirshahi, U.; Murray, M.F.; Smelser, D.T.; Gerhard, G.S.; Ledbetter, D.H. The Geisinger MyCode community health initiative: An electronic health record–linked biobank for precision medicine research. Genet. Med. 2016, 18, 906–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.L.; McCormick, C.Z.; Lazzeri, A.L.; Lindbuchler, D.M.; Hallquist, M.L.; Manickam, K.; Buchanan, A.H.; Rahm, A.K.; Giovanni, M.A.; Frisbie, L.; et al. A Model for Genome-First Care: Returning Secondary Genomic Findings to Participants and Their Healthcare Providers in a Large Research Cohort. Am. J. Hum. Genet. 2018, 103, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivakumar, M.; Miller, J.E.; Dasari, V.R.; Gogoi, R.P.; Kim, D. Exome-Wide Rare Variant Analysis from the DiscovEHR Study Identifies Novel Candidate Predisposition Genes for Endometrial Cancer. Front. Oncol. 2019, 9, 574. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, E.; Katsila, T.; Mitropoulou, C.; Tsermpini, E.-E.; Patrinos, G.P. Integrating Next-Generation Sequencing in the Clinical Pharmacogenomics Workflow. Front. Pharmacol. 2019, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Mizzi, C.; Peters, B.A.; Mitropoulou, C.; Mitropoulos, K.; Katsila, T.; Agarwal, M.R.; Van Schaik, R.H.; Drmanac, R.; Borg‡, J.; Patrinos, G.P. Personalized pharmacogenomics profiling using whole-genome sequencing. Pharmacogenomics 2014, 15, 1223–1234. [Google Scholar] [CrossRef]
- Mizzi, C.; Dalabira, E.; Kumuthini, J.; Dzimiri, N.; Balogh, I.; Başak, N.; Böhm, R.; Borg, J.; Borgiani, P.; Božina, N.; et al. A European Spectrum of Pharmacogenomic Biomarkers: Implications for Clinical Pharmacogenomics. PLoS ONE 2016, 11, e0162866. [Google Scholar] [CrossRef]
- Tasa, T.; Krebs, K.; Kals, M.; Mägi, R.; Lauschke, V.M.; Haller, T.; Puurand, T.; Remm, M.; Esko, T.; Metspalu, A.; et al. Genetic variation in the Estonian population: Pharmacogenomics study of adverse drug effects using electronic health records. Eur. J. Hum. Genet. 2018, 27, 442–454. [Google Scholar] [CrossRef] [Green Version]
- Bomba, L.; Walter, K.; Soranzo, N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017, 18, 77. [Google Scholar] [CrossRef]
- Kryukov, G.; Pennacchio, L.A.; Sunyaev, S.R. Most Rare Missense Alleles Are Deleterious in Humans: Implications for Complex Disease and Association Studies. Am. J. Hum. Genet. 2007, 80, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Tennessen, J.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.R.; Wegmann, D.; Ehm, M.G.; Kessner, D.; Jean, P.S.; Verzilli, C.; Shen, J.; Tang, Z.; Bacanu, S.-A.; Fraser, D.; et al. An Abundance of Rare Functional Variants in 202 Drug Target Genes Sequenced in 14,002 People. Science 2012, 337, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikura, K.; Ingelman-Sundberg, M.; Lauschke, V.M. Genetic variation in the human cytochrome P450 supergene family. Pharmacogenet. Genom. 2015, 25, 1–594. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Exome Aggregation Consortium; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Lauschke, V.M.; Milani, L.; Ingelman-Sundberg, M. Pharmacogenomic Biomarkers for Improved Drug Therapy—Recent Progress and Future Developments. AAPS J. 2017, 20. [Google Scholar] [CrossRef]
- Lakiotaki, K.; Kanterakis, A.; Kartsaki, E.; Katsila, T.; Patrinos, G.P.; Potamias, G. Exploring public genomics data for population pharmacogenomics. PLoS ONE 2017, 12, e0182138. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Eilbeck, K.; Lewis, S.; Mungall, C.; Yandell, M.; Stein, L.; Durbin, R.; Ashburner, M. The Sequence Ontology: A tool for the unification of genome annotations. Genome Biol. 2005, 6, R44. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; Montgomery, S.B.; et al. A Systematic Survey of Loss-of-Function Variants in Human Protein-Coding Genes. Science 2012, 335, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Arbitrio, M.; Di Martino, M.T.; Scionti, F.; Agapito, G.; Guzzi, P.H.; Cannataro, M.; Tassone, P.; Tagliaferri, P. DMET™ (Drug Metabolism Enzymes and Transporters): A pharmacogenomic platform for precision medicine. Oncotarget 2016, 7, 54028–54050. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2016, 19, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspar, S.; Schneider, T.; Meienberg, J.; Matyas, G. Added Value of Clinical Sequencing: WGS-Based Profiling of Pharmacogenes. Int. J. Mol. Sci. 2020, 21, 2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacogene Family | VEP Consequence | Novel/Previously Known |
|---|---|---|
| Phase I Metabolizing Enzymes | missense_variant: 214 missense_variant,splice_region_variant: 10 | Novel: 69 Known: 155 |
| Phase II Metabolizing Enzymes | missense_variant: 80 missense_variant,splice_region_variant: 5 | Novel: 19 Known: 66 |
| Transporters | missense_variant: 369 missense_variant,splice_region_variant: 9 | Novel: 125 Previously known: 253 |
| Others | missense_variant: 113 missense_variant,splice_region_variant: 4 | Novel: 32 Previously known: 85 |
| Pharmacogene Category | N | GENES | IMPACT |
|---|---|---|---|
| ENZ I | 188 | CYP4B1, DPYD, CYP2C19, CYP2C9, CYP2C8, CYP2E1, CYP1A1, CYP1A2, CYP4F2, CYP2A6, CYP2B6, CYP2A13, CYP2F1, CYP2S1, CYP1B1, CYP2D6, CYP39A1, CYP3A5, CYP3A7, CYP3A4, CYP3A43 | HIGH: 11 LOW: 10 MODERATE: 158 MODIFIER: 9 |
| ENZ II | 115 | SULT2A1, SULT1C2, UGT1A8, UGT1A10, UGT1A6, COMT, UGT2B15, TPMT, NAT1, NAT2 | HIGH: 2 LOW: 18 MODERATE: 54 MODIFIER: 41 |
| TRANSPORTERS | 22 | ABCC2, SLCO1B1, SLC22A1, ABCB1 | HIGH: 0 LOW: 5 MODERATE: 17 MODIFIER: 0 |
| OTHERS | 8 | CDA, VKORC1, G6PD | HIGH: 0 LOW: 2 MODERATE: 5 MODIFIER: 1 |
| Consequence | Number of Variants per Gene | Number of Variants per Functionality |
|---|---|---|
| frameshift_variant | CYP2D6: 2 CYP3A5: 1 | No: 3 |
| frameshift_variant,splice_region_variant | CYP2C9: 1 | No: 1 |
| inframe_deletion,splice_region_variant | CYP2D6: 1 | Decreased: 1 |
| intron_variant | CYP2D6: 2 DPYD: 1 UGT1A6: 2 | Normal: 1 Decreased: 3 No: 1 |
| missense_variant | CYP2B6: 4 CYP2C19: 3 CYP2C9: 7 CYP2D6: 2 CYP4F2: 1 DPYD:43 SLCO1B1: 5 TPMT: 4 | Normal: 36 Possibly Decreased: 7 Decreased: 9 No: 17 |
| missense_variant,splice_region_variant | DPYD:1 | Normal: 1 |
| splice_acceptor_variant | CYP2D6: 1 TPMT: 2 | No: 3 |
| splice_donor_variant | DPYD: 1 | No: 1 |
| splice_region_variant, intron_variant | DPYD: 1 | Normal: 1 |
| splice_region_variant, synonymous_variant | DPYD: 1 | Normal: 1 |
| start_lost | TPMT: 1 | No: 1 |
| synonymous_variant | CYP3A5: 1 DPYD: 3 | Normal: 3 No: 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandi, M.-T.; Williams, M.S.; van der Spek, P.; Koromina, M.; Patrinos, G.P. Exome-Wide Analysis of the DiscovEHR Cohort Reveals Novel Candidate Pharmacogenomic Variants for Clinical Pharmacogenomics. Genes 2020, 11, 561. https://doi.org/10.3390/genes11050561
Pandi M-T, Williams MS, van der Spek P, Koromina M, Patrinos GP. Exome-Wide Analysis of the DiscovEHR Cohort Reveals Novel Candidate Pharmacogenomic Variants for Clinical Pharmacogenomics. Genes. 2020; 11(5):561. https://doi.org/10.3390/genes11050561
Chicago/Turabian StylePandi, Maria-Theodora, Marc S. Williams, Peter van der Spek, Maria Koromina, and George P. Patrinos. 2020. "Exome-Wide Analysis of the DiscovEHR Cohort Reveals Novel Candidate Pharmacogenomic Variants for Clinical Pharmacogenomics" Genes 11, no. 5: 561. https://doi.org/10.3390/genes11050561
APA StylePandi, M. -T., Williams, M. S., van der Spek, P., Koromina, M., & Patrinos, G. P. (2020). Exome-Wide Analysis of the DiscovEHR Cohort Reveals Novel Candidate Pharmacogenomic Variants for Clinical Pharmacogenomics. Genes, 11(5), 561. https://doi.org/10.3390/genes11050561