Distinct Tumor Microenvironments Are a Defining Feature of Strain-Specific CRISPR/Cas9-Induced MPNSTs

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. CRISPR/Cas9 Generated MPNSTs and Growth Analysis
2.3. Generation of Cell Lines from MPNSTs
2.4. Indel Analysis
2.5. Histology and Immunohistochemistry
2.6. Quantitative RT-PCR
2.7. Statistical Analysis
3. Results
3.1. Host Strain Determines Tumor Onset for Genetically-Identical MPNSTs
3.2. Indel Analysis Reveals Unique Patterns of Gene Disruption
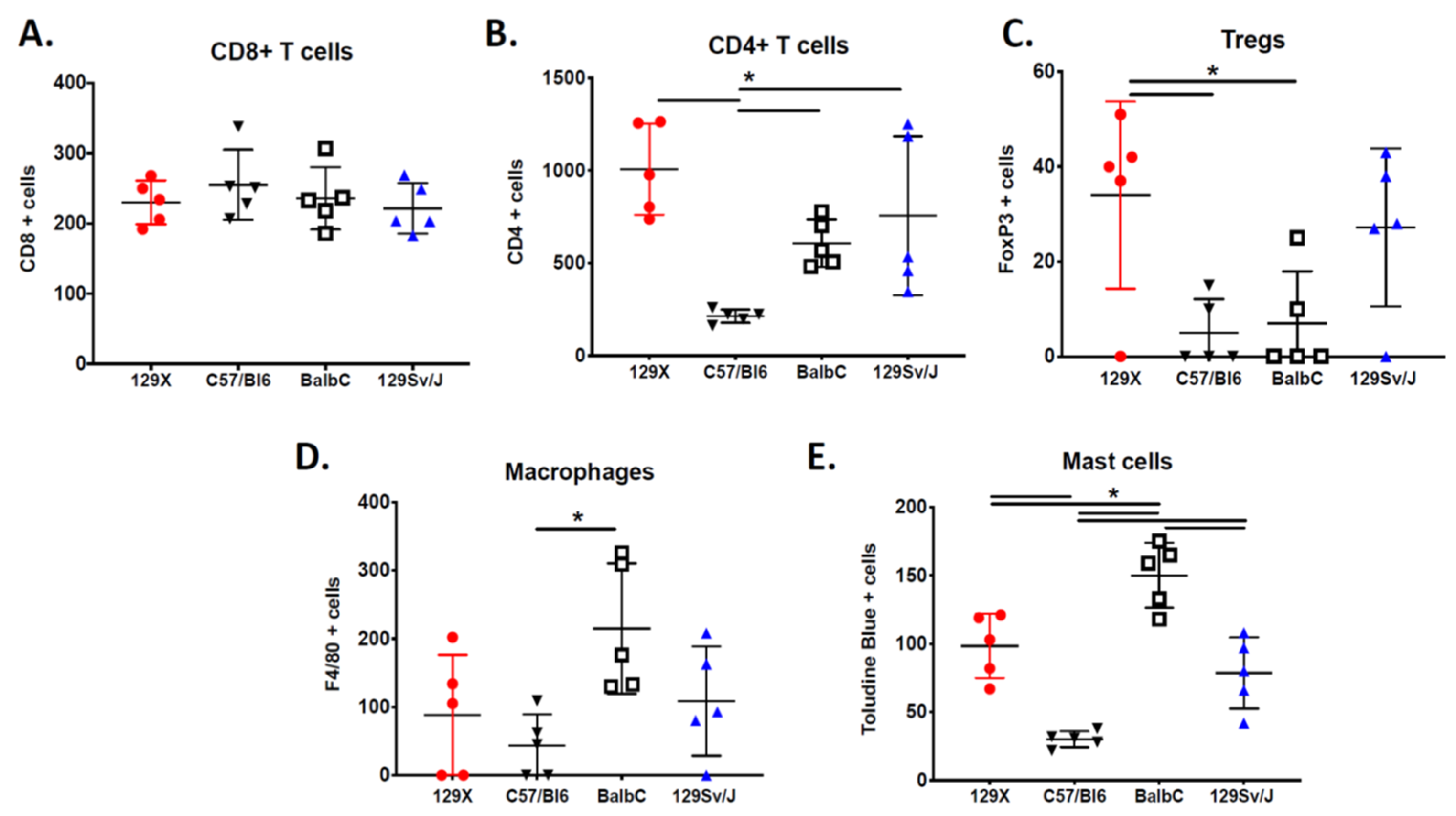
3.3. Immunological Diversity of MPNSTs Is a Hallmark of Genetic Background
3.4. Gene Expression of the MPNST Microenvironment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reilly, K.M. The Effects of Genetic Background of Mouse Models of Cancer: Friend or Foe? Cold Spring Harb. Protoc. 2016, 2016, pdb.top076273. [Google Scholar] [CrossRef] [PubMed]
- Kuperwasser, C.; Hurlbut, G.D.; Kittrell, F.S.; Dickinson, E.S.; Laucirica, R.; Medina, D.; Naber, S.P.; Jerry, D.J. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am. J. Pathol. 2000, 157, 2151–2159. [Google Scholar] [CrossRef]
- Koch, J.G.; Gu, X.; Han, Y.; El-Naggar, A.K.; Olson, M.V.; Medina, D.; Jerry, D.J.; Blackburn, A.C.; Peltz, G.; Amos, C.I.; et al. Mammary tumor modifiers in BALB/cJ mice heterozygous for p53. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2007, 18, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, A.C.; Hill, L.Z.; Roberts, A.L.; Wang, J.; Aud, D.; Jung, J.; Nikolcheva, T.; Allard, J.; Peltz, G.; Otis, C.N.; et al. Genetic mapping in mice identifies DMBT1 as a candidate modifier of mammary tumors and breast cancer risk. Am. J. Pathol. 2007, 170, 2030–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, L.P.; Albers, J.; Hejhal, T.; Pfundstein, S.; Gonçalves, A.F.; Catalano, A.; Wild, P.J.; Frew, I.J. Mouse genetic background influences whether HrasG12V expression plus Cdkn2a knockdown causes angiosarcoma or undifferentiated pleomorphic sarcoma. Oncotarget 2018, 9, 19753–19766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragani, T.A. 10 years of mouse cancer modifier loci: Human relevance. Cancer Res. 2003, 63, 3011–3018. [Google Scholar]
- Dietrich, W.F.; Lander, E.S.; Smith, J.S.; Moser, A.R.; Gould, K.A.; Luongo, C.; Borenstein, N.; Dove, W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell 1993, 75, 631–639. [Google Scholar] [CrossRef]
- Reilly, K.M.; Loisel, D.A.; Bronson, R.T.; McLaughlin, M.E.; Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 2000, 26, 109–113. [Google Scholar] [CrossRef]
- Reilly, K.M.; Tuskan, R.G.; Christy, E.; Loisel, D.A.; Ledger, J.; Bronson, R.T.; Smith, C.D.; Tsang, S.; Munroe, D.J.; Jacks, T. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc. Natl. Acad. Sci. USA 2004, 101, 13008–13013. [Google Scholar] [CrossRef] [Green Version]
- Reilly, K.M.; Broman, K.W.; Bronson, R.T.; Tsang, S.; Loisel, D.A.; Christy, E.S.; Sun, Z.; Diehl, J.; Munroe, D.J.; Tuskan, R.G. An imprinted locus epistatically influences Nstr1 and Nstr2 to control resistance to nerve sheath tumors in a neurofibromatosis type 1 mouse model. Cancer Res. 2006, 66, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Brosius, S.N.; Turk, A.N.; Byer, S.J.; Brossier, N.M.; Kohli, L.; Whitmire, A.; Mikhail, F.M.; Roth, K.A.; Carroll, S.L. Neuregulin-1 overexpression and Trp53 haploinsufficiency cooperatively promote de novo malignant peripheral nerve sheath tumor pathogenesis. Acta Neuropathol. (Berl.) 2014, 127, 573–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-L.; Xu, P.-Z.; Peng, X.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 2006, 20, 1569–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Lifsted, T.; Le Voyer, T.; Williams, M.; Muller, W.; Klein-Szanto, A.; Buetow, K.H.; Hunter, K.W. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int. J. Cancer 1998, 77, 640–644. [Google Scholar] [CrossRef]
- Sellers, R.S.; Clifford, C.B.; Treuting, P.M.; Brayton, C. Immunological variation between inbred laboratory mouse strains: Points to consider in phenotyping genetically immunomodified mice. Vet. Pathol. 2012, 49, 32–43. [Google Scholar] [CrossRef]
- Hensel, J.A.; Khattar, V.; Ashton, R.; Ponnazhagan, S. Characterization of immune cell subtypes in three commonly used mouse strains reveals gender and strain-specific variations. Lab. Investig. J. Technol. Methods Pathol. 2019, 99, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Harrison, D.E. Quantitative trait loci regulating relative lymphocyte proportions in mouse peripheral blood. Blood 2002, 99, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. Baltim. Md 1950 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Foerster, F.; Boegel, S.; Heck, R.; Pickert, G.; Rüssel, N.; Rosigkeit, S.; Bros, M.; Strobl, S.; Kaps, L.; Aslam, M.; et al. Enhanced protection of C57 BL/6 vs. Balb/c mice to melanoma liver metastasis is mediated by NK cells. Oncoimmunology 2018, 7, e1409929. [Google Scholar] [CrossRef] [Green Version]
- White, P.; Liebhaber, S.A.; Cooke, N.E. 129 × 1/SvJ mouse strain has a novel defect in inflammatory cell recruitment. J. Immunol. Baltim. Md 1950 2002, 168, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.D.; Carter, K.J.; Jean-Philippe, S.R.; Chang, M.; Mobashery, S.; Thiolloy, S.; Lynch, C.C.; Matrisian, L.M.; Fingleton, B. Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 2008, 68, 6251–6259. [Google Scholar] [CrossRef] [Green Version]
- Mosely, S.I.S.; Prime, J.E.; Sainson, R.C.A.; Koopmann, J.-O.; Wang, D.Y.Q.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.W.; Bhattacharya, S.; Yanamandra, N.; Kilian, D.; Shi, H.; Yadavilli, S.; Katlinskaya, Y.; Kaczynski, H.; Conner, M.; Benson, W.; et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS ONE 2018, 13, e0206223. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. (Hagerstown Md.: 1997) 2013, 36, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasselly, C.; Denis, M.; Bourguignon, A.; Talhi, N.; Mathe, D.; Tourette, A.; Serre, L.; Jordheim, L.P.; Matera, E.L.; Dumontet, C. The Antitumor Activity of Combinations of Cytotoxic Chemotherapy and Immune Checkpoint Inhibitors Is Model-Dependent. Front. Immunol. 2018, 9, 2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, R.; Neri, D. Potentiation of PD-L1 blockade with a potency-matched dual cytokine-antibody fusion protein leads to cancer eradication in BALB/c-derived tumors but not in other mouse strains. Cancer Immunol. Immunother. CII 2018, 67, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, M.; Whitley, M.J.; Kuo, H.-C.; Xu, E.S.; Walens, A.; Mowery, Y.M.; Van Mater, D.; Eward, W.C.; Cardona, D.M.; et al. Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat. Commun. 2017, 8, 15999. [Google Scholar] [CrossRef] [PubMed]
- Dodd, R.D.; Añó, L.; Blum, J.M.; Li, Z.; Van Mater, D.; Kirsch, D.G. Methods to generate genetically engineered mouse models of soft tissue sarcoma. Methods Mol. Biol. Clifton NJ 2015, 1267, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Dodd, R.D.; Mito, J.K.; Eward, W.C.; Chitalia, R.; Sachdeva, M.; Ma, Y.; Barretina, J.; Dodd, L.; Kirsch, D.G. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol. Cancer Ther. 2013, 12, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- Dodd, R.D.; Lee, C.-L.; Overton, T.; Huang, W.; Eward, W.C.; Luo, L.; Ma, Y.; Ingram, D.R.; Torres, K.E.; Cardona, D.M.; et al. NF1+/- Hematopoietic Cells Accelerate Malignant Peripheral Nerve Sheath Tumor Development without Altering Chemotherapy Response. Cancer Res. 2017, 77, 4486–4497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat. Commun. 2016, 7, 10770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synthego. ICE v2 CRISPR Analysis Tools. Available online: https://www.synthego.com/products/bioinformatics/crispr-analysis (accessed on 3 January 2020).
- Lindeboom, R.G.H.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, K.T.; Li, L.S.; Kim, N.-G.; Kang, H.J.; Koh, K.H.; Chwae, Y.-J.; Kim, K.M.; Kim, Y.K.; Park, S.M.; Jang, S.K.; et al. Selective Translational Repression of Truncated Proteins from Frameshift Mutation-Derived mRNAs in Tumors. PLoS Biol. 2007, 5, e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staser, K.; Yang, F.-C.; Clapp, D.W. Mast cells and the neurofibroma microenvironment. Blood 2010, 116, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoni, A.; Colombo, M.P.; Pucillo, C. The Role of Mast Cells in Molding the Tumor Microenvironment. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2015, 8, 167–176. [Google Scholar] [CrossRef] [Green Version]
- de Vasconcelos, R.A.T.; Guimarães Coscarelli, P.; Vieira, T.M.; Noguera, W.S.; Rapozo, D.C.M.; Acioly, M.A. Prognostic significance of mast cell and microvascular densities in malignant peripheral nerve sheath tumor with and without neurofibromatosis type 1. Cancer Med. 2019, 8, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, M.; Koyanagi, M.; Arimura, Y. Comparative Analysis of Bone Marrow-derived Mast Cell Differentiation in C57BL/6 and BALB/c Mice. Immunol. Investig. 2019, 48, 303–320. [Google Scholar] [CrossRef]
- Noguchi, J.; Kuroda, E.; Yamashita, U. Strain difference of murine bone marrow-derived mast cell functions. J. Leukoc. Biol. 2005, 78, 605–611. [Google Scholar] [CrossRef]
- Pae, S.; Cho, J.Y.; Dayan, S.; Miller, M.; Pemberton, A.D.; Broide, D.H. Chronic allergen challenge induces bronchial mast cell accumulation in BALB/c but not C57BL/6 mice and is independent of IL-9. Immunogenetics 2010, 62, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.J.; Farber, C.R.; Orozco, L.; Kang, H.M.; Ghazalpour, A.; Siemers, N.; Neubauer, M.; Neuhaus, I.; Yordanova, R.; Guan, B.; et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010, 20, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscher, K.; Ehinger, E.; Gupta, P.; Pramod, A.B.; Wolf, D.; Tweet, G.; Pan, C.; Mills, C.D.; Lusis, A.J.; Ley, K. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat. Commun. 2017, 8, 16041. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001, 2, 731–737. [Google Scholar] [CrossRef]
- Mock, B.A.; Krall, M.M.; Dosik, J.K. Genetic mapping of tumor susceptibility genes involved in mouse plasmacytomagenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 9499–9503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Ramsay, E.S.; Mock, B.A. Cdkn2a, the cyclin-dependent kinase inhibitor encoding p16INK4a and p19ARF, is a candidate for the plasmacytoma susceptibility locus, Pctr1. Proc. Natl. Acad. Sci. USA 1998, 95, 2429–2434. [Google Scholar] [CrossRef] [Green Version]
- Sittig, L.J.; Carbonetto, P.; Engel, K.A.; Krauss, K.S.; Barrios-Camacho, C.M.; Palmer, A.A. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 2016, 91, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Tsang, J.S.; Schwartzberg, P.L.; Kotliarov, Y.; Biancotto, A.; Xie, Z.; Germain, R.N.; Wang, E.; Olnes, M.J.; Narayanan, M.; Golding, H.; et al. Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell 2014, 157, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Casanova, J.-L.; Abel, L. The human model: A genetic dissection of immunity to infection in natural conditions. Nat. Rev. Immunol. 2004, 4, 55–66. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scherer, A.; Stephens, V.R.; McGivney, G.R.; Gutierrez, W.R.; Laverty, E.A.; Knepper-Adrian, V.; Dodd, R.D. Distinct Tumor Microenvironments Are a Defining Feature of Strain-Specific CRISPR/Cas9-Induced MPNSTs. Genes 2020, 11, 583. https://doi.org/10.3390/genes11050583
Scherer A, Stephens VR, McGivney GR, Gutierrez WR, Laverty EA, Knepper-Adrian V, Dodd RD. Distinct Tumor Microenvironments Are a Defining Feature of Strain-Specific CRISPR/Cas9-Induced MPNSTs. Genes. 2020; 11(5):583. https://doi.org/10.3390/genes11050583
Chicago/Turabian StyleScherer, Amanda, Victoria R. Stephens, Gavin R. McGivney, Wade R. Gutierrez, Emily A. Laverty, Vickie Knepper-Adrian, and Rebecca D. Dodd. 2020. "Distinct Tumor Microenvironments Are a Defining Feature of Strain-Specific CRISPR/Cas9-Induced MPNSTs" Genes 11, no. 5: 583. https://doi.org/10.3390/genes11050583
APA StyleScherer, A., Stephens, V. R., McGivney, G. R., Gutierrez, W. R., Laverty, E. A., Knepper-Adrian, V., & Dodd, R. D. (2020). Distinct Tumor Microenvironments Are a Defining Feature of Strain-Specific CRISPR/Cas9-Induced MPNSTs. Genes, 11(5), 583. https://doi.org/10.3390/genes11050583