Simultaneous Detection of NF1, SPRED1, LZTR1, and NF2 Gene Mutations by Targeted NGS in an Italian Cohort of Suspected NF1 Patients

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. DNA and RNA Extraction and Retro-Transcription
2.3. Multiplex Ligation-Dependent Probe Amplification Analysis (MLPA)
2.4. NGS Sequencing
2.5. Data Analysis
2.6. Sanger Sequencing
2.7. Submission of Genomic Variations
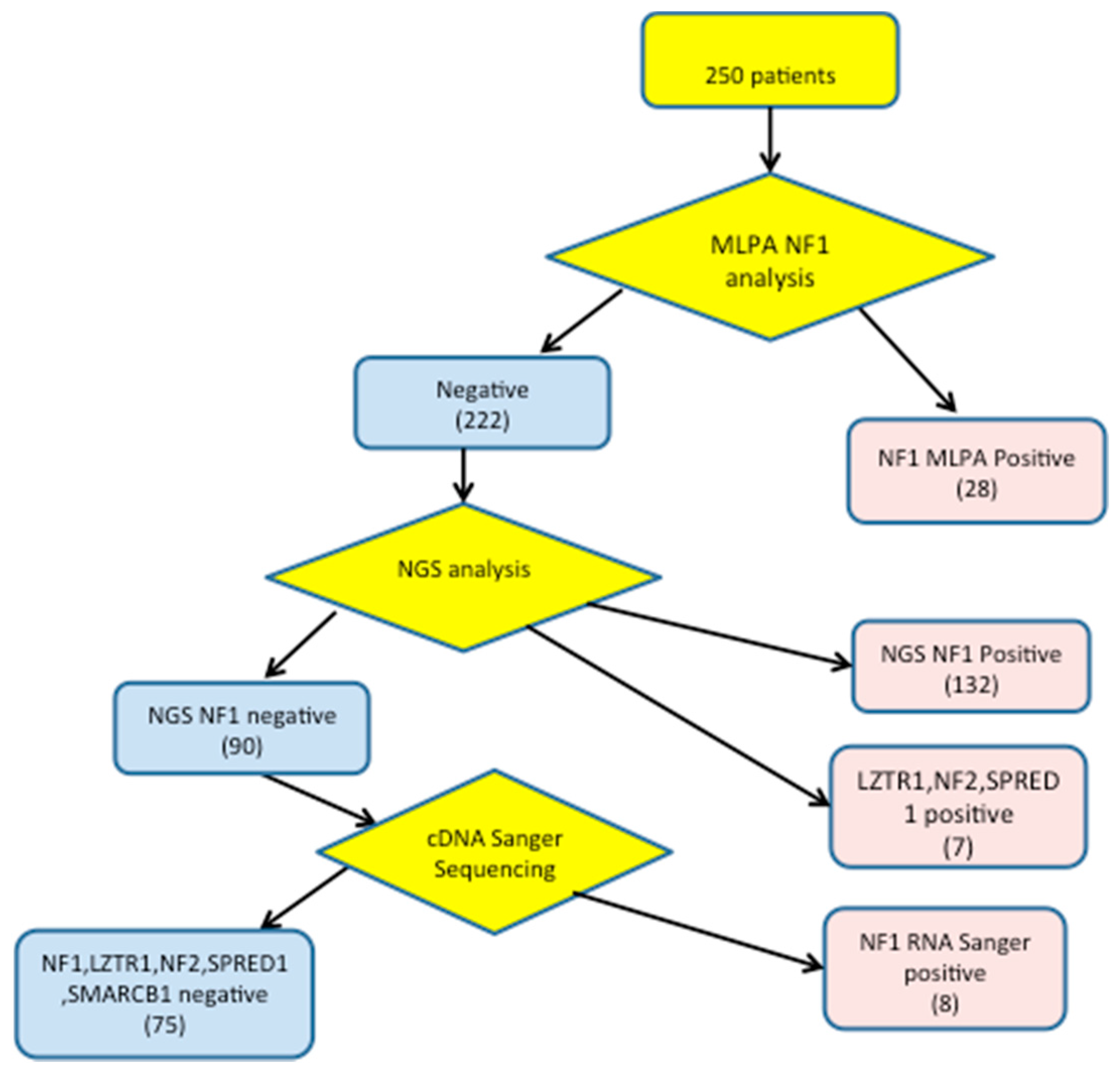
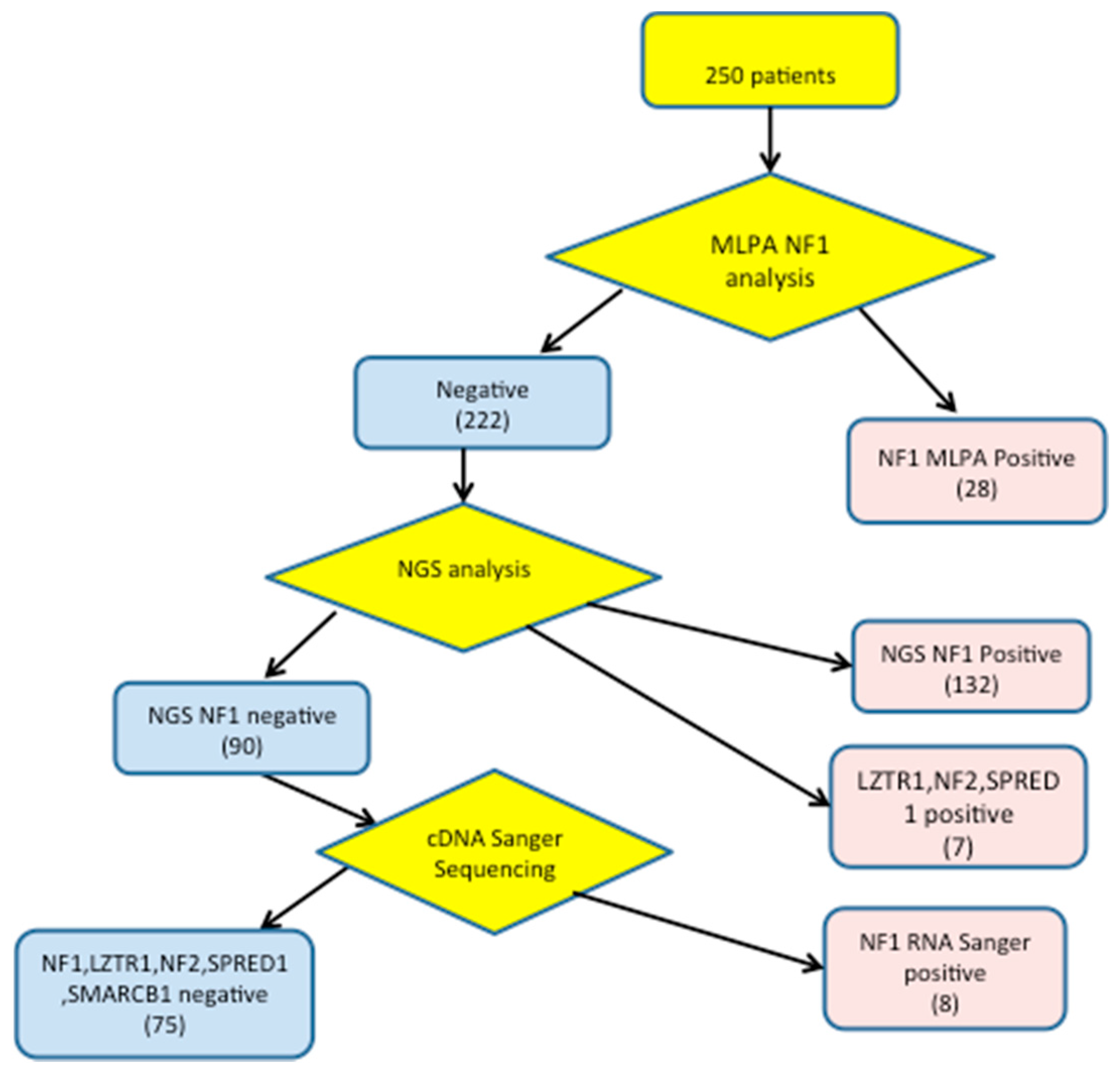
3. Results
3.1. Detection of NF1 Pathogenic Variants
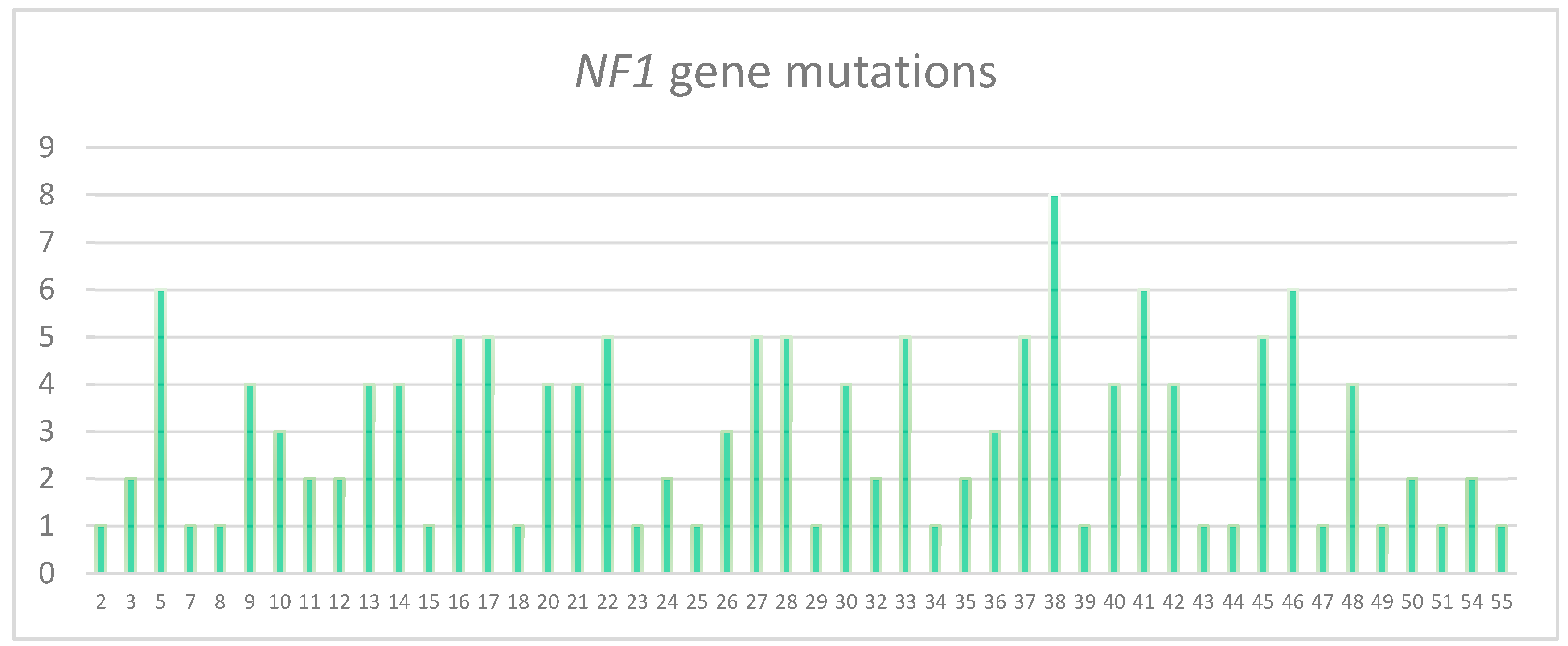
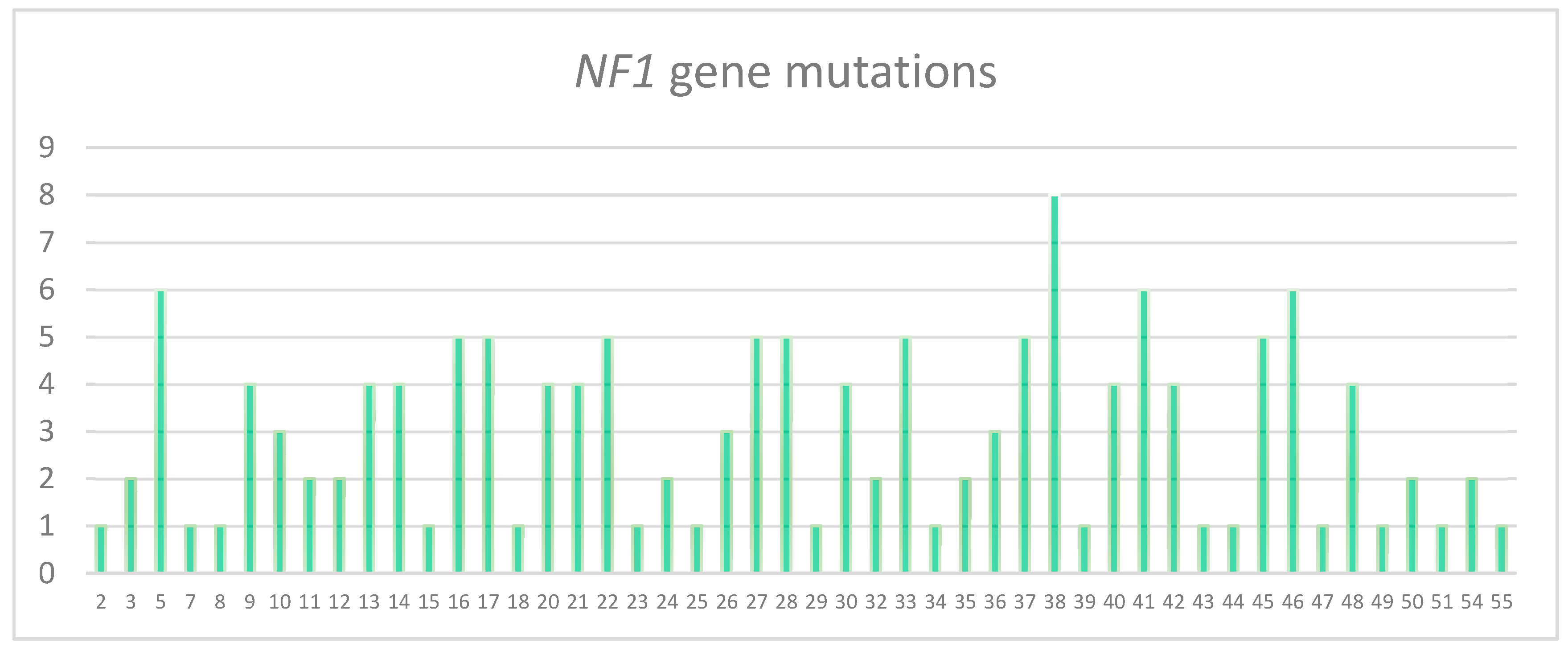
3.2. Characterization of the Pathogenic Variants Detected by NGS
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Cawthon, R.M.; Weiss, R.; Xu, G.F.; Viskochil, D.; Culver, M.; Stevens, J.; Robertson, M.; Dunn, D.; Gesteland, R.; O’Connell, P. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990, 62, 193–201. [Google Scholar] [CrossRef]
- Huson, S.M. The Neurofibromatoses: Classification, Clinical Features and Genetic Councelling. In Neurofibromatoses; Kaufmann, D., Ed.; Karger Publishers: Basel, Switzerland, 2008; pp. 1–20. [Google Scholar]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Wimmer, K.; Roca, X.; Beiglbock, H.; Callens, T.; Etzler, J.; Rao, A.R.; Krainer, A.R.; Fonatsch, C.; Messiaen, L. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5’ splice-site disruption. Hum. Mutat. 2007, 28, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, L.; Siebert, R.; Gesk, S.; Friedrich, R.E.; Tinschert, S.; Kehrer-Sawatzki, H.; Mautner, V.F. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum. Mutat. 2004, 23, 111–116. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Bengesser, K.; Callens, T.; Mikhail, F.; Fu, C.; Hillmer, M.; Walker, M.E.; Saal, H.M.; Lacassie, Y.; Cooper, D.N.; et al. Identification of large NF1 duplications reciprocal to NAHR-mediated type-1 NF1 deletions. Hum. Mut. 2014, 35, 1469–1475. [Google Scholar] [CrossRef]
- Van Minkelen, R.; van Bever, Y.; Kromosoeto, J.N.R.; Withagen-Hermans, C.J.; Nieuwlaat, A.; Halley, D.J.J.; van den Ouweland, A.M.W. A clinical and genetic overview of 18 years neurofibromatosis type 1 molecular diagnostics in the Netherlands. Clin. Genet. 2014, 85, 318–327. [Google Scholar] [CrossRef]
- Evans, D.G.; Bowers, N.; Burkitt-Wright, E.; Miles, E.; Garg, S.; Scott-Kitching, V.; Penman-Splitt, M.; Dobbie, A.; Howard, E.; Ealing, J.; et al. Comprehensive RNA analysis of the NF1 gene in classically affected NF1 affected individuals meeting NIH criteria has high sensitivity and mutation negative testing is reassuring in isolated cases with pigmentary features only. EBioMedicine 2016, 7, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Hutter, S.; Piro, R.M.; Waszak, S.M.; Kehrer-Sawatzki, H.; Friedrich, R.E.; Lassaletta, A.; Witt, O.; Korbel, J.O.; Lichter, P.; Schuhmann, M.U.; et al. No correlation between NF1 mutation position and risk of optic pathway glioma in 77 unrelated NF1 patients. Hum. Genet. 2016, 135, 469–475. [Google Scholar] [CrossRef]
- Thomas, L.; Spurlock, G.; Eudall, C.; Thomas, N.S.; Mort, M.; Hamby, S.E.; Chuzhanova, N.; Brems, H.; Legius, E.; Cooper, D.N.; et al. Exploring the somatic NF1 mutational spectrum associated with NF1 cutaneous neurofibromas. Eur. J. Hum. Genet. 2012, 20, 411–419. [Google Scholar] [CrossRef] [Green Version]
- National Institutes of Health Consensus Development Panel. Neurofibromatosis: Conference Statement. Arch. Neurol. 1988, 45, 575–578. [Google Scholar] [CrossRef]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; de Schepper, S.; Fryns, J.P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis1-like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messiaen, L.; Yao, S.; Brems, H.; Callens, T.; Sathienkijkanchai, A.; Denayer, E.; Spencer, E.; Arn, P.; Babovic-Vuksanovic, D.; Bay, C.; et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA 2009, 302, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, M.; Polizzi, A.; Spalice, A.; Salpietro, V.; Caltabiano, R.; D’Orazi, V.; Pavone, P.; Pirrone, C.; Magro, G.; Platania, N.; et al. The natural history of spinal neurofibromatosis: A critical review of clinical and genetic features. Clin. Genet. 2015, 87, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Giugliano, T.; Melone, M.A.B.; Cirillo, M.; Schettino, C.; Bernardo, P.; Cirillo, G.; Perrotta, S.; Piluso, G. Multiple spinal nerve enlargement and SOS1 mutation: Further evidence of overlap between neurofibromatosis type 1 and Noonan phenotype. Clin. Genet. 2018, 93, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Rosenbaum, T.; Messiaen, L. Connection between constitutional mismatch repair deficiency 2 syndrome and Neurofibromatosis type 1. Clin. Genet. 2017, 91, 507–519. [Google Scholar] [CrossRef]
- Bianchessi, D.; Morosini, S.; Saletti, V.; Ibba, M.C.; Natacci, F.; Esposito, S.; Cesaretti, C.; Riva, D.; Finocchiaro, G.; Eoli, M. 126 novel mutations in Italian patients with neurofibromatosis type 1. Mol. Genet. Genom. Med. 2015, 3, 513–525. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; Dunnen, J.T.d. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice Site Detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human SplicingFinder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.J.; Zhang, C.; Wang, J.; Chew, S.L.; Zhang, M.Q.; Krainer, A.R. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 2006, 15, 2490–2508. [Google Scholar] [CrossRef] [Green Version]
- Seong, M.W.; Yeo, I.K.; Cho, S.I.; Park, C.K.; Kim, S.K.; Paek, S.H.; Kim, D.G.; Jung, H.W.; Park, H.; Kim, S.Y.; et al. Molecular characterization of the NF2 gene in Korean patients with neurofibromatosis type 2: A report of four novel mutations. Korean J. Lab. Med. 2010, 30, 190–194. [Google Scholar] [CrossRef]
- Tsipi, M.; Poulou, M.; Fylaktou, I.; Kosma, K.; Tsoutsou, E.; Pons, M.R.; Kokkinou, E.; Kitsiou-Tzeli, S.; Fryssira, H.; Tzetis, M. Phenotypic expression of a spectrum of Neurofibromatosis Type 1 (NF1) mutations identified through NGS and MLPA. J. Neurol. Sci. 2018, 395, 95–105. [Google Scholar] [CrossRef]
- Calì, F.; Chiavetta, V.; Ruggeri, G.; Piccione, M.; Selicorni, A.; Palazzo, D.; Bonsignore, M.; Cereda, A.; Elia, M.; Failla, P.; et al. Mutation spectrum of NF1 gene in Italian patients with neurofibromatosis type 1 using Ion Torrent PGM™ platform. Eur. J. Med. Genet. 2017, 60, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Giugliano, T.; Santoro, C.; Torella, A.; del Vecchio, F.; Grandone, B.A.; Onore, M.E.; Melone, M.R.B.; Straccia, G.; Melis, D.; Piccolo, V. Clinical and Genetic Findings in Children with Neurofibromatosis Type 1, Legius Syndrome, and Other Related Neurocutaneous Disorders. Genes 2019, 10, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu-Chou, Y.H.; Hung, T.C.; Lin, Y.T.; Cheng, H.W.; Lin, H.W.; Lin, C.H.; Yu, C.C.; Chen, K.T.; Yeh, T.H.; Chen, Y.R. Genetic diagnosis of neurofibromatosis type 1: Targeted next- generation sequencing with Multiple Ligation-Dependent Probe Amplification analysis. J. Biomed. Sci. 2018, 25, 72. [Google Scholar] [CrossRef] [PubMed]
- Ulusal, S.D.; Gürkan, H.; Atlı, E.; Özal, S.A.; Çiftdemir, M.; Tozkır, H.; Karal, Y.; Gü lü, H.; Eker, D.; Görker, I. Genetic analyses of the NF1 gene in Turkish neurofibromatosis type 1 patients and definition of three novel variants. BJMG 2017, 20, 13–20. [Google Scholar] [PubMed]
- Toliat, M.R.; Erdogan, F.; Gewies, A.; Fahsold, R.; Buske, A.; Tinschert, S.; Nürnberg, P. Analysis of the NF1 gene by temperature gradient gel electrophoresisreveals a high incidence of mutations in exon 4b. Electrophoresis 2000, 21, 541–544. [Google Scholar] [CrossRef]
- Ruggieri, M.; Huson, S.M. The clinical and diagnostic implications of mosaicismin the neurofibromatoses. Neurology 2001, 56, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Ogose, A.; Hotta, T.; Imaizumi, S.; Saito, H.; Homma, T.; Takahashi, H.E. Deepseated segmental neurofibromatosis without cafe au lait spots. Skelet. Radiol. 2000, 29, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; de Schepper, S.; Vandesompele, B.; Heyns, I.; Janssens, S.; Speleman, F.; Legius, E.; Messiaen, L. Molecular dissection of isolated disease features in mosaic neurofibromatosis type 1. Am. J. Hum. Genet. 2007, 81, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, S.; Thompson, P.; Frayling, I.; Upadhyaya, M. Molecular diagnosis of neurofibromatosis type 1: 2 years experience. Fam. Cancer 2007, 6, 21–34. [Google Scholar] [CrossRef]
- Pacot, L.; Burin des Roziers, C.; Laurendeau, I.; Briand-Suleau, A.; Coustier, A.; Mayard, T.; Tlemsani, C.; Faivre, L.; Thomas, Q.; Rodriguez, T. One NF1 Mutation may Conceal Another. Genes 2019, 10, 633. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.R.; Blakeley, J.O.; Evans, D.G.; Hanemann, C.O.; Hulsebos, T.J.; Hunter-Schaedle, K.; Kalpana, G.V.; Korf, B.; Messiaen, L.; Papi, L.; et al. Update from the 2011International Schwannomatosis Workshop: From genetics to diagnostic criteria. Am. J. Med. Genet. 2013, 161, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Piotrowski, A.; Xie, J.; Liu, Y.F.; Poplawski, A.B.; Gomes, A.R.; Madanecki, P.; Fu, C.; Crowley, M.R.; Crossman, D.K.; Armstrong, L.; et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 2014, 46, 182–187. [Google Scholar] [CrossRef]
- Smith, M.; Kulkarni, A.; Rustad, C.; Bowers, N.L.; Wallace, A.J.; Holder, S.E.; Heiberg, A.; Ramsden, R.T.; Evans, D.G. Vestibular schwannoma occur in schwannomatosis and should not be considered an exclusion criteria for clinical diagnosis. Am. J. Med. Genet. 2012, 158, 215–219. [Google Scholar] [CrossRef]
- Smith, M.J.; Bulman, M.; Gokhale, C.; Wallace, A.J.; King, A.T.; Lloyd, S.K.; Rutherford, S.A.; Hammerbeck-Ward, C.L.; Freeman, S.R.; Evans, D.G. Revisiting neurofibromatosis type 2 diagnostic criteria to exclude LZTR1 related schwannomatosis. Neurology 2017, 88, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, G.L.; Aguena, M.; Gos, M.; Hung, C.; Pilch, S.; Fahiminiya, S.; Abramowicz, A.; Cristian, I.; Buscarilli, M.; Naslavsky, M.S.; et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J. Med. Genet. 2015, 52, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquinet, A.; Bonnard, A.; Capri, Y.; Martin, D.; Sadzot, B.; Bianchi, E.; Servais, L.; Sacré, J.P.; Cavé, H.; Verloes, A. Oligo-astrocytoma in LZTR1-related noonan syndrome. Eur. J. Med. Genet. 2019, 18, 30629–30633. [Google Scholar] [CrossRef] [PubMed]
- Deiller, A.C.; Van-Gilsa, J.; Zordana, C.; Tinata, J.; Loiseauc, H.; Fabred, T.; Dellecie, C.; Cohenf, J.; Vidaudf, M.; Parfaitf, M.; et al. Coexistence of schwannomatosis and glioblastoma in two families. Eur. J. Med. Genet. 2019, 62, 103680. [Google Scholar] [CrossRef]
- Motta, M.; Fidan, M.; Bellacchio, E.; Pantaleoni, F.; Schneider-Heieck, K.; Coppola, S.; Borck, G.; Salviati, L.; Zenker, M.; Cirstea, I.C.; et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum. Mol. Genet. 2019, 28, 1007–1022. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Variant On Genome/DBID. | DNA Change | RNA Change | Protein Change |
|---|---|---|---|
| NF1_002811 | c.980delT | r.980delu | p.Leu327Argfs*49 |
| NF1_002809 | c.269T>G | r.269u>g | p.Leu90Arg |
| NF1_000806 | c.6755A>G | r.6642_6756del115 | p.Phe2215fs |
| NF1_002823 | c.6770dupG | r.6770dupg | p.Cys2257Trpfs*6 |
| NF1_002817 | c.3574G>T | r.3574g>u | p.Glu1192* |
| NF1_002812 | c.1170delC | r.1170delc | p.Asn390Lysfs*22 |
| NF1_002816 | c.2026_2027insT | r.2026_2027insu | p.Thr676fs |
| NF1_002810 | c.586+2 T>G | r.480_586del107 | p.Leu161fs |
| NF1_002819 | c.4653delT | r.4653delu | p.Phe1551Leufs*2 |
| NF1_001963 | c.5870T>C | r.5870u>c | p.Leu1957Pro |
| NF1_002826 | c.7907+1 _7907+4 delGTAA | r.7807_7907del101 | p.Thr2604* |
| NF1_002818 | c.3703C>T | r.3703c>u | p.Gln1235* |
| NF1_002815 | c.1541_1542delAG | r.1541_1542delag | p.Gln514Argfs*43 |
| NF1_002813 | c.1185+2delT | r.1063_1185del123 | p.Asn355_Lys395del |
| NF1_002821 | c.5199_5205delTATTAAA | r.5199_5205deluauuaaa | p.Ile1734Leufs*8 |
| NF1_002822 | c.6756+11 C>T | r.6642_6756del115 | p.Phe2215Hisfs*6 |
| NF1_002822 | c.6756+11 C>T | r.6642_6756del115 | p.Phe2215Hisfs*6 |
| NF1_002824 | c.7345_7346delAA | r.7345_7346delaa | p.Asn2449Cysfs*12 |
| NF1_002814 | c.1393-3 delTA | r.1393_1527del135 | p.Ser465_Cys509del |
| NF1_002825 | c.7433dupG | r.7433dupg | p.Ser2479fs |
| NF1_002820 | c.4773-2A>C | r.4773_5065del293 | p.Phe1592Leufs*7 |
| NF1_002834 | c.6326_6329delTTCA | r.6326_6329deluuca | p.Ile2109Thrfs*19 |
| NF1_002832 | c.4701_4708delAACGTTAA | r.4701_4708delAaacguuaa | p.Thr1568Tyrfs*30 |
| NF1_002827 | c.288+1137C>T | r.288_289ins288+1019_288+1136 ins118 | p.Gly96Glu97ins39+fs*10 |
| NF1_002829 | c.3197+2T>A | r.3114_3197del84 | p.Arg1038_Thr1066del |
| NF1_002828 | c.2810T>A | r.2810u>a | p.Leu937* |
| NF1_002831 | c.4684G>T | r.4684g>u | p.Glu1562* |
| NF1_002830 | c.3314+1G>C | r.3275_3314del40 | p.Gly1092Aspfs*7 |
| NF1_002833 | c.5425delC | r.5425delc | p.Arg1809Alafs*33 |
| NF1_002301 | c.2915T>C | r.2915u>c | p.Leu972Pro |
| NF1_002841 | c.5513_5514delTA | r.5513_5514delua | p.Leu1838Serfs*2 |
| NF1_002836 | c.1280delC | r.1280delc | p.Pro427Leufs*46 |
| NF1_002838 | c.3564_3565delACinsTGA | r.3564_3565delacinsuga | p.Gln1188Hisfs*7 |
| NF1_002840 | c.4719_4720dupAC | r.4719_4720dupac | p.Gln1574fs |
| NF1_002842 | c.5989A>C | r.5989a>c | p.Ser1997Arg |
| NF1_002837 | c.3315-8 T>G | r.3315_3496del182 | p.Thr1106Leufs*28 |
| NF1_002844 | c.7921dupG | r.7921dupg | p.Asp2641fs |
| NF1_002301 | c.2915T>C | r.2915u>c | p.Leu972Pro |
| NF1_002839 | c.3709-9T>A | r.3708_3709insuucucag | p.Asp1237Phefs*4 |
| NF1_002835 | c.1260+2 T>G | r.1260_1261insggaaguccaaaag | p.Ser421Glyfs*12 |
| NF1_002843 | c.6085G>T | r.6085_6364del280 | p.Val2029Lysfs*7 |
| NF1_002861 | c.-383_(60+1_61-1)del | r.(?) | p.(?) |
| NF1_001695 | c.(3708+1_3709-1)_(3974+1_3975-1)dup | r.(?) | p.(?) |
| NF1_002858 | c.4435A>G | r.4368_4435del68 | p.Phe1457* |
| NF1_002862 | c.1944_1945delAGinsC | r.1944_1945delinsc | p.Glu648Aspfs*40 |
| NF1_002858 | c.1122_1125delTCTA | r.1122_1125delucua | p.Asp374Glufs*2 |
| NF1_002858 | c.6762_6783delTGAGAGTTGCTTAAAAGGACCT | r.6762_6783del22 | p.Glu2255Thrfs*8 |
| NF1_002860 | c.1463_1466dupGCTA | r.1463_1466dupgcua | p.Tyr489* |
| NF1_002859 | c.7151_7161delTTGTTGCAAGA | r.7151_7161del | p.Ile2384Asnfs*13 |
| NF1_002858 | c.4435A>G | r.4368_4435del68 | p.Phe1457* |
| NF1_002864 | c.6005T>A | r.6005u>a | p.Leu2002* |
| NF1_002863 | c.7000-?_8314+?del | r.7000_8314del1314 | p.Ser2334Glufs*59 |
| LZTR1_000102 | c.844C>T | r.844c>u | p.Gln282* |
| LZTR1_000103 | c.154delC | r.154delc | p.His52Ilefs*49 |
| LZTR1_000041 | c.1394C>T | r.1394c>u | p.Ala465Val |
| LZTR1_000104 | c.161G>A | r.161g>a | p.Trp54* |
| Mutation Type | Location | n°pts |
|---|---|---|
| c.288 + 1138 C>T | Intron 3 | 1 |
| c.499_502 del TGTT | Exon 5 | 3 |
| c.1021_1022 del GT | Exon 9 | 1 |
| c.2033 dupC | Exon 18 | 1 |
| c.4224_4225 del AA insT | Exon 32 | 1 |
| c.7907 + 791 C>G | Intron 54 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchessi, D.; Ibba, M.C.; Saletti, V.; Blasa, S.; Langella, T.; Paterra, R.; Cagnoli, G.A.; Melloni, G.; Scuvera, G.; Natacci, F.; et al. Simultaneous Detection of NF1, SPRED1, LZTR1, and NF2 Gene Mutations by Targeted NGS in an Italian Cohort of Suspected NF1 Patients. Genes 2020, 11, 671. https://doi.org/10.3390/genes11060671
Bianchessi D, Ibba MC, Saletti V, Blasa S, Langella T, Paterra R, Cagnoli GA, Melloni G, Scuvera G, Natacci F, et al. Simultaneous Detection of NF1, SPRED1, LZTR1, and NF2 Gene Mutations by Targeted NGS in an Italian Cohort of Suspected NF1 Patients. Genes. 2020; 11(6):671. https://doi.org/10.3390/genes11060671
Chicago/Turabian StyleBianchessi, Donatella, Maria Cristina Ibba, Veronica Saletti, Stefania Blasa, Tiziana Langella, Rosina Paterra, Giulia Anna Cagnoli, Giulia Melloni, Giulietta Scuvera, Federica Natacci, and et al. 2020. "Simultaneous Detection of NF1, SPRED1, LZTR1, and NF2 Gene Mutations by Targeted NGS in an Italian Cohort of Suspected NF1 Patients" Genes 11, no. 6: 671. https://doi.org/10.3390/genes11060671
APA StyleBianchessi, D., Ibba, M. C., Saletti, V., Blasa, S., Langella, T., Paterra, R., Cagnoli, G. A., Melloni, G., Scuvera, G., Natacci, F., Cesaretti, C., Finocchiaro, G., & Eoli, M. (2020). Simultaneous Detection of NF1, SPRED1, LZTR1, and NF2 Gene Mutations by Targeted NGS in an Italian Cohort of Suspected NF1 Patients. Genes, 11(6), 671. https://doi.org/10.3390/genes11060671