Genome-Wide Identification of Discriminative Genetic Variations in Beef and Dairy Cattle via an Information-Theoretic Approach


Abstract
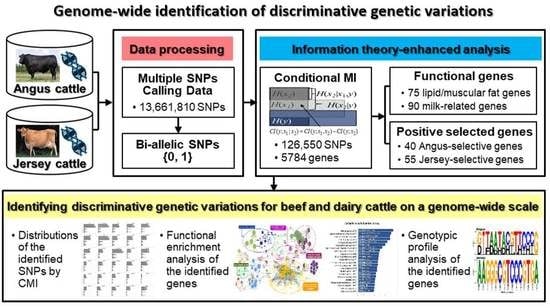
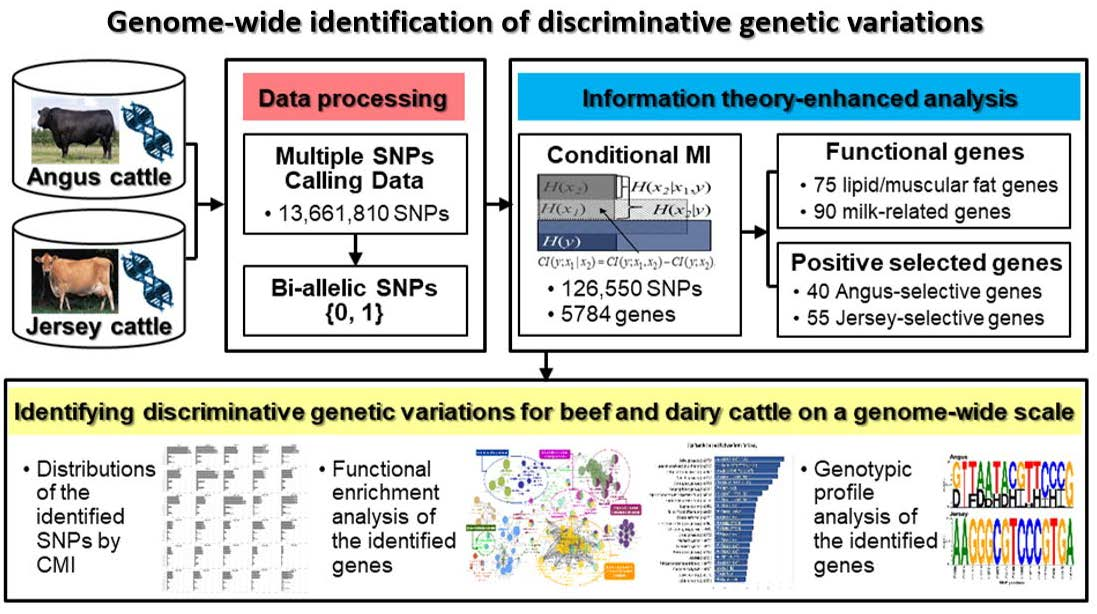
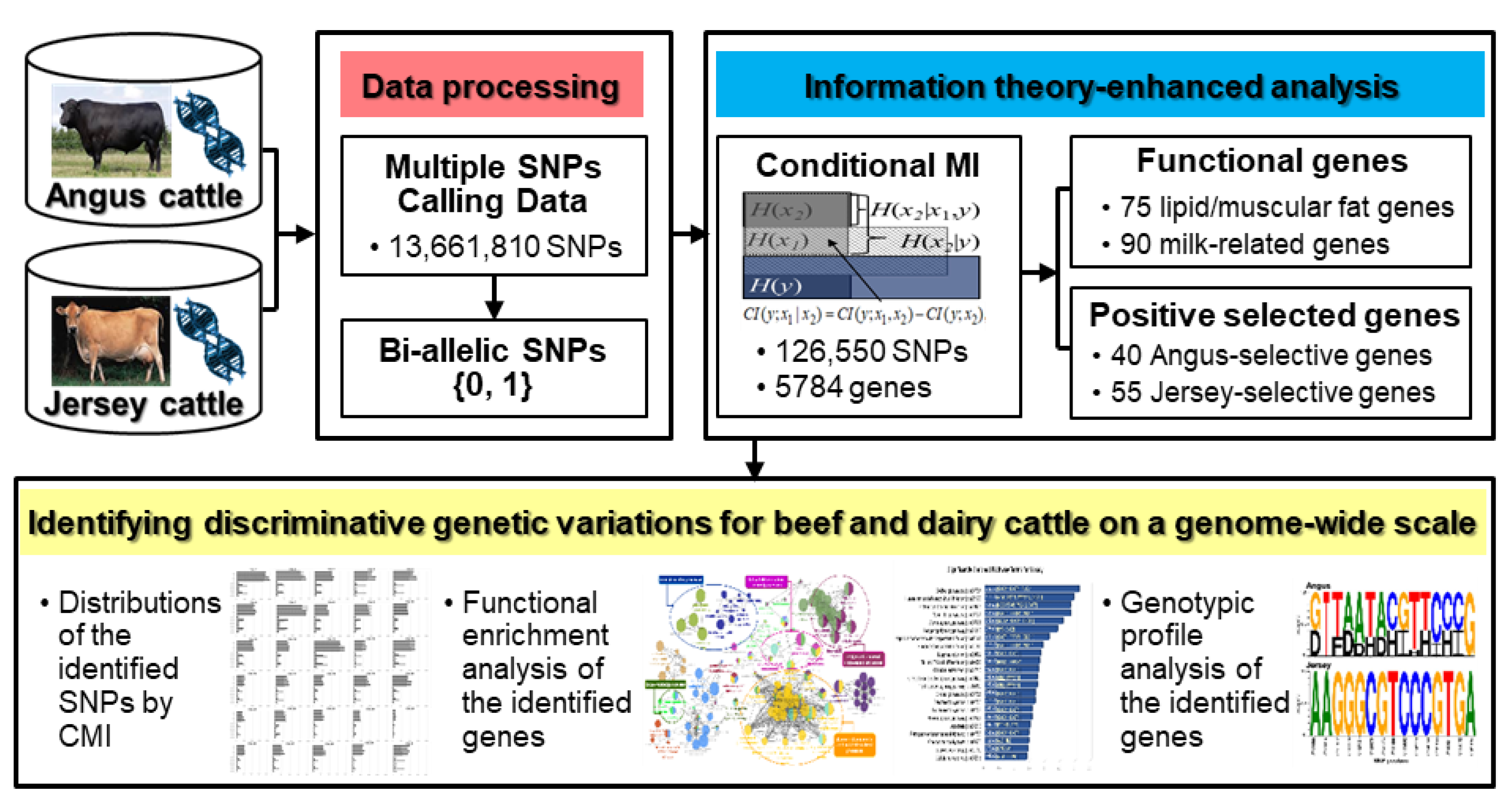
1. Introduction
2. Materials and Methods
2.1. Sequencing, Quality Control, and Variant Calling
2.2. Conditional Mutual Information
2.3. XP-CLR and XP-EHH Tests
3. Results
3.1. SNP Detection
3.2. Population Structures
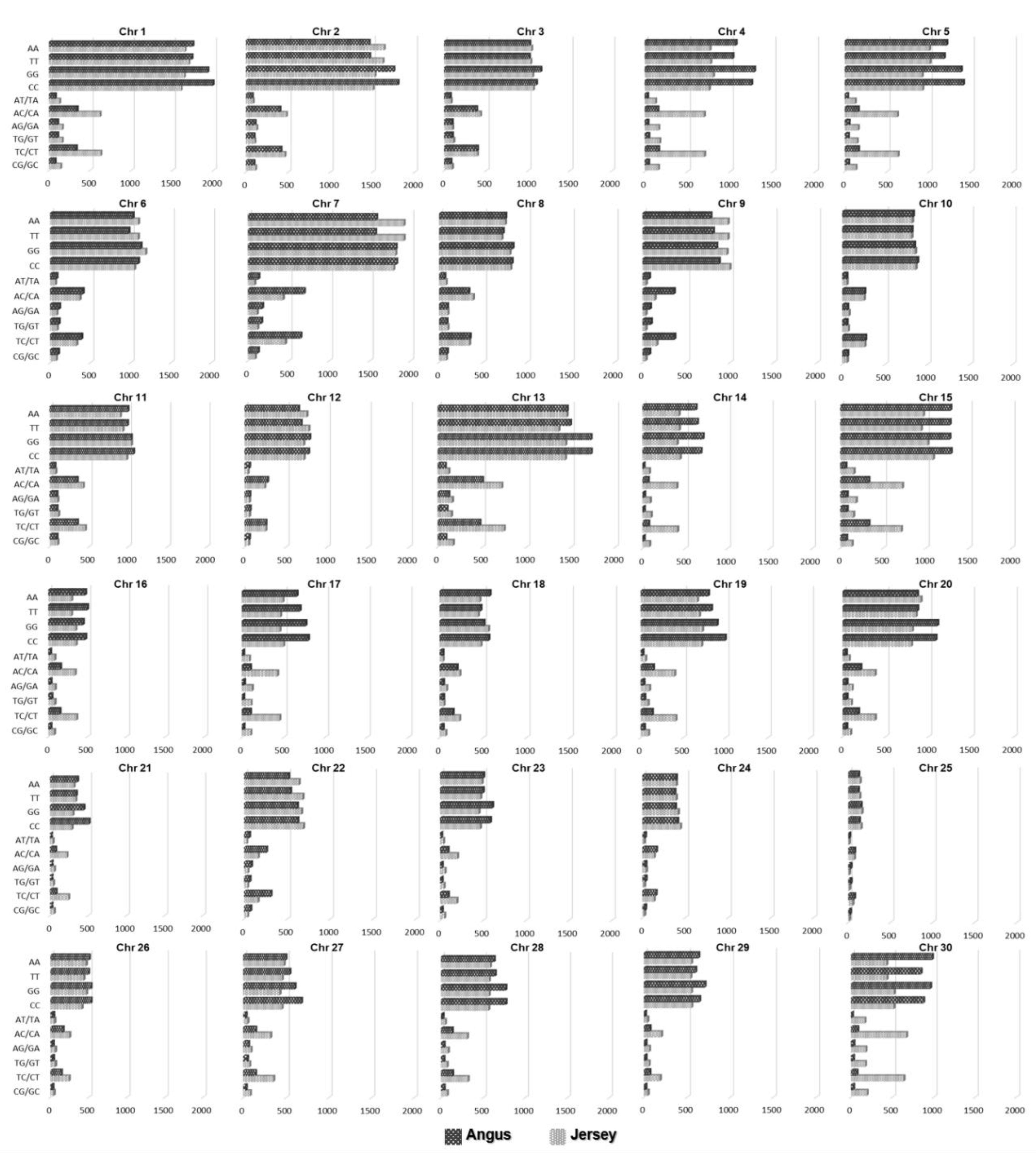
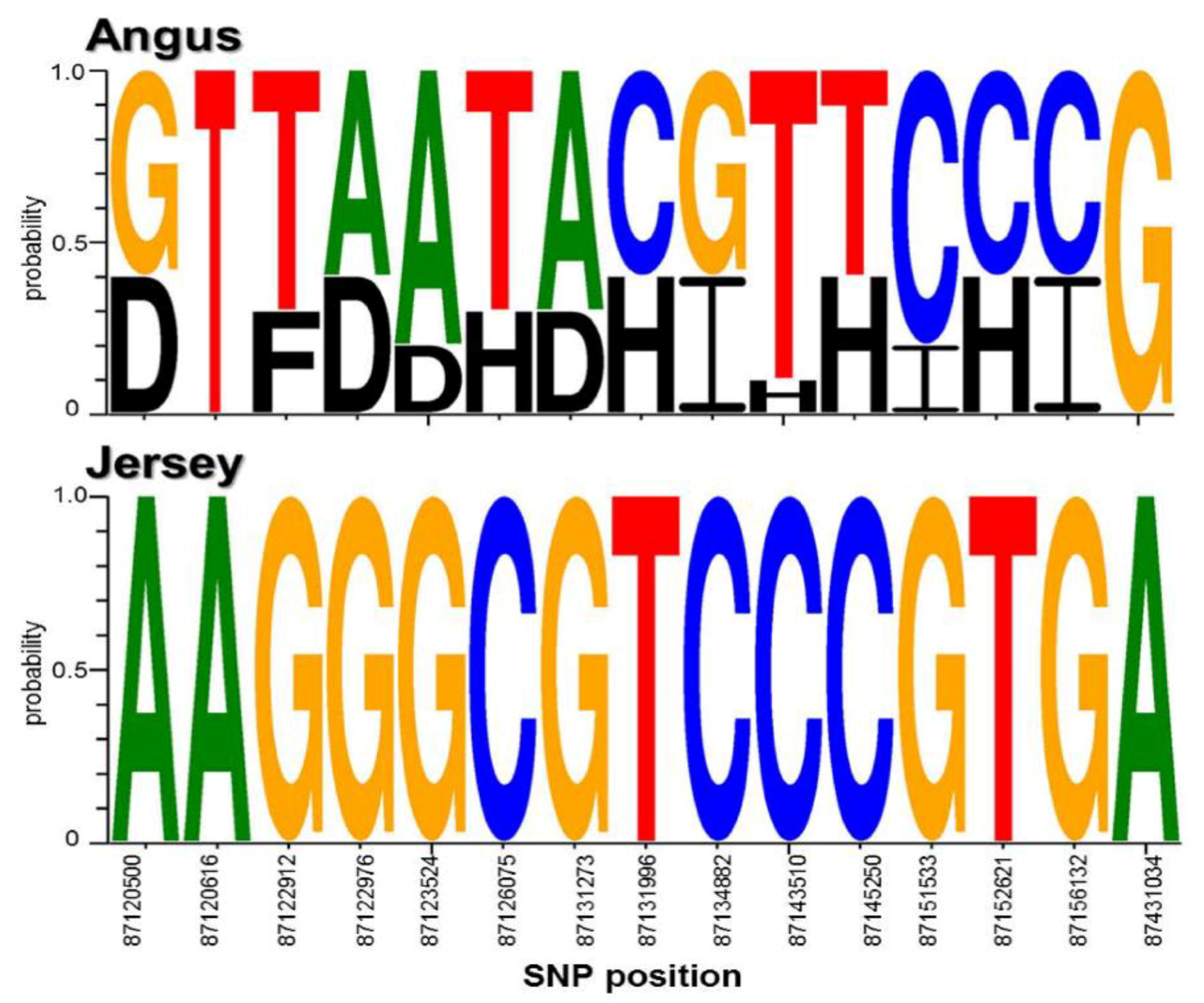
3.3. Extraction of Discriminative SNPs Based on the Information-Theoretic Method
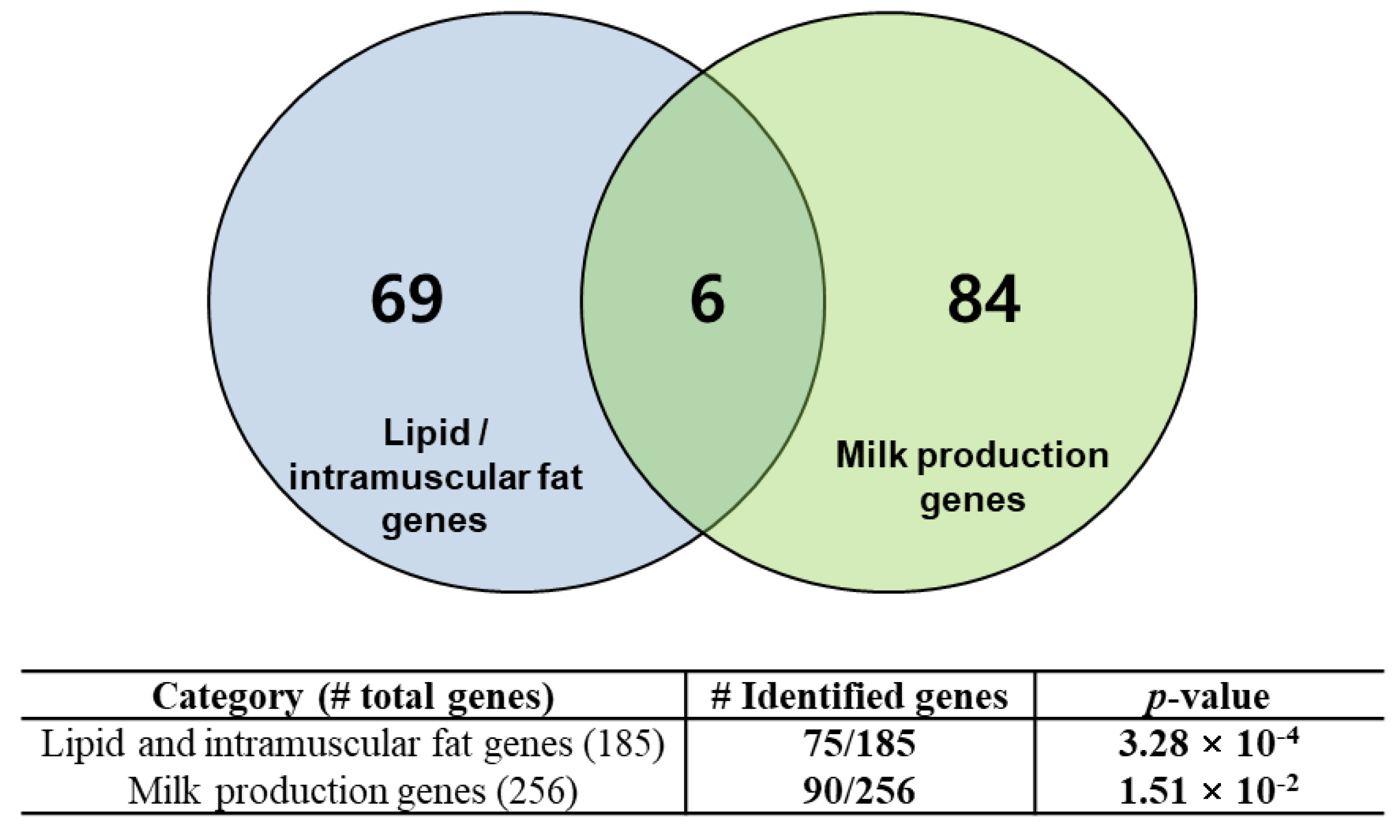
3.4. Identification of Breed-Specific Genes
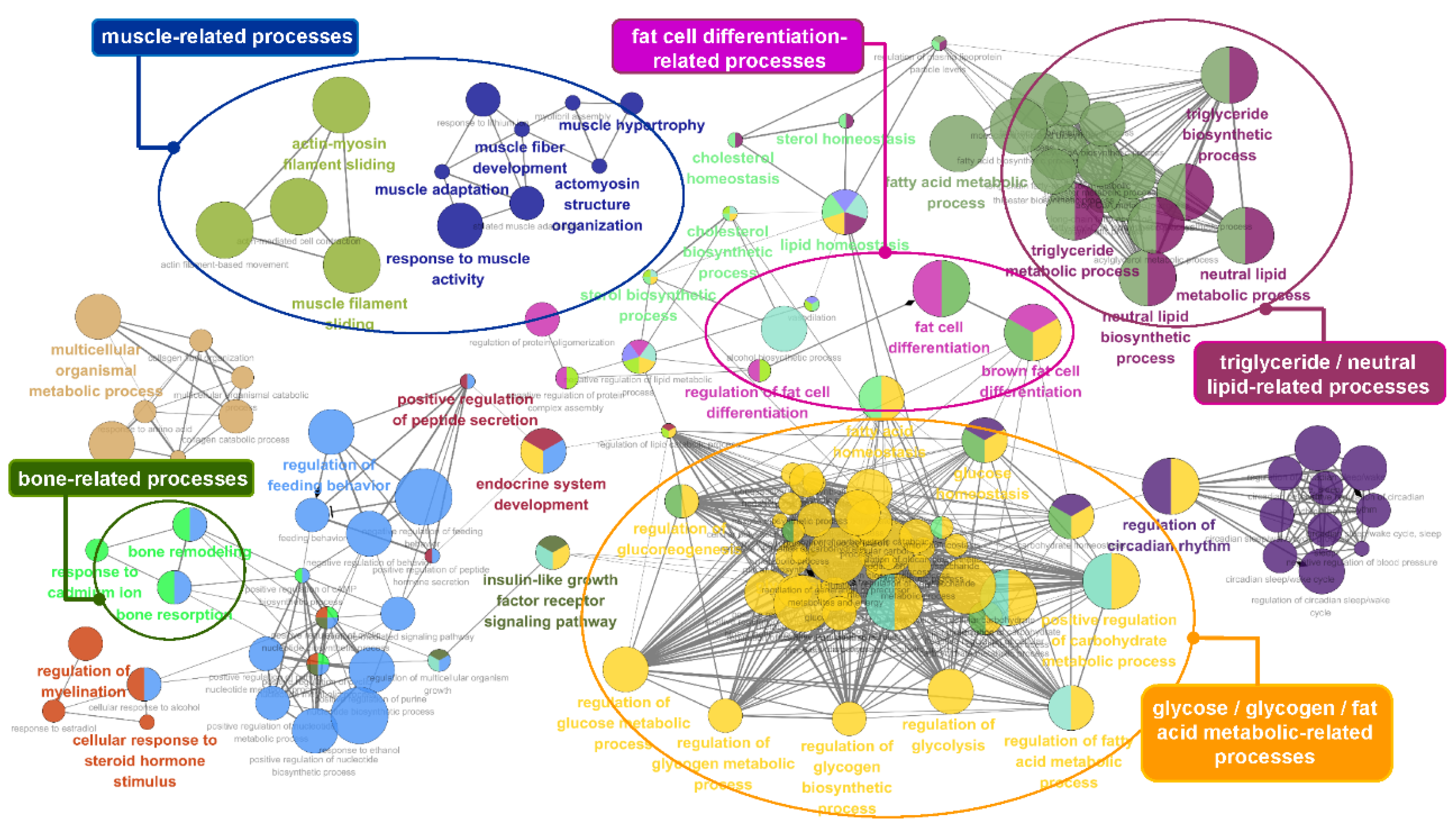
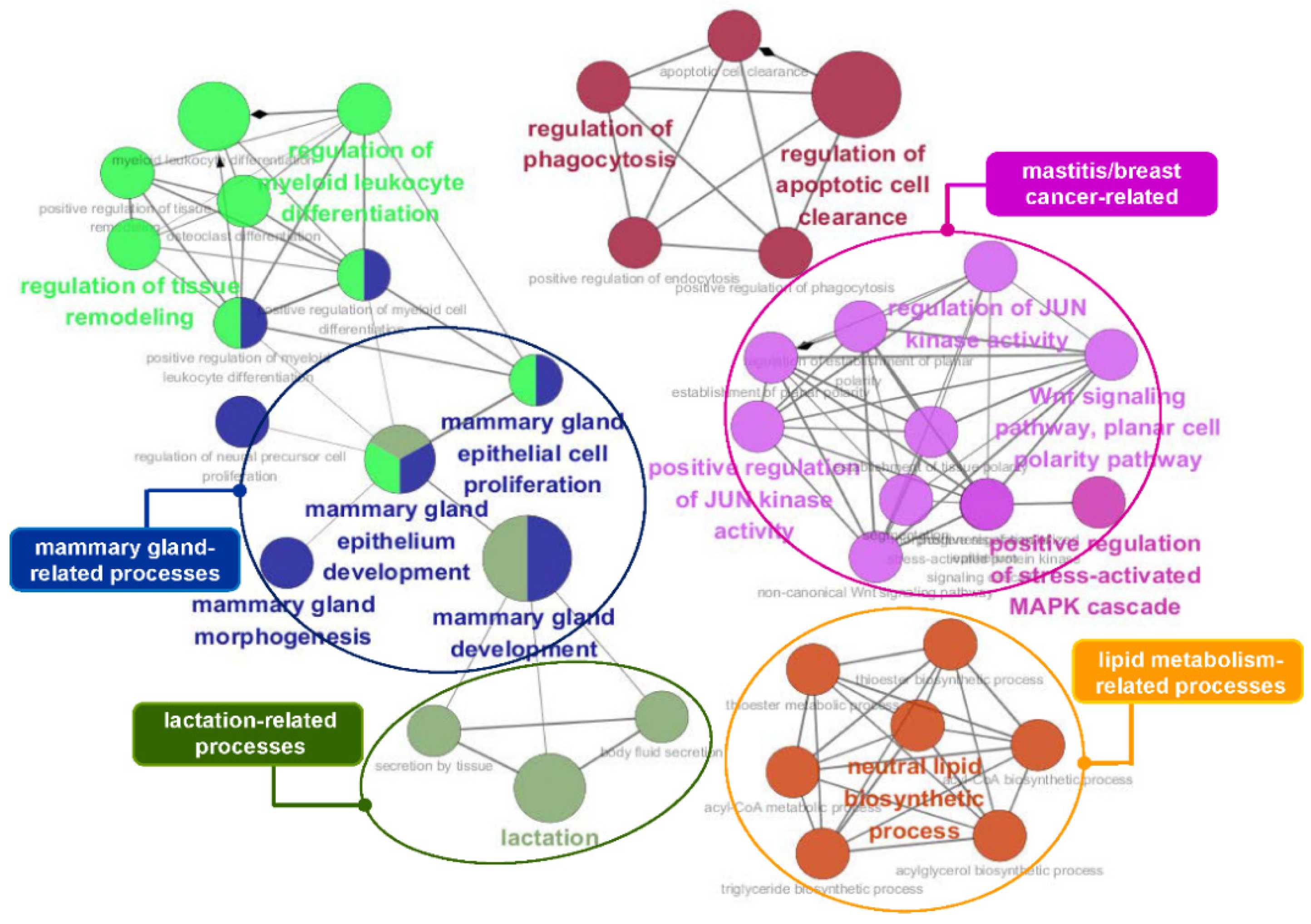
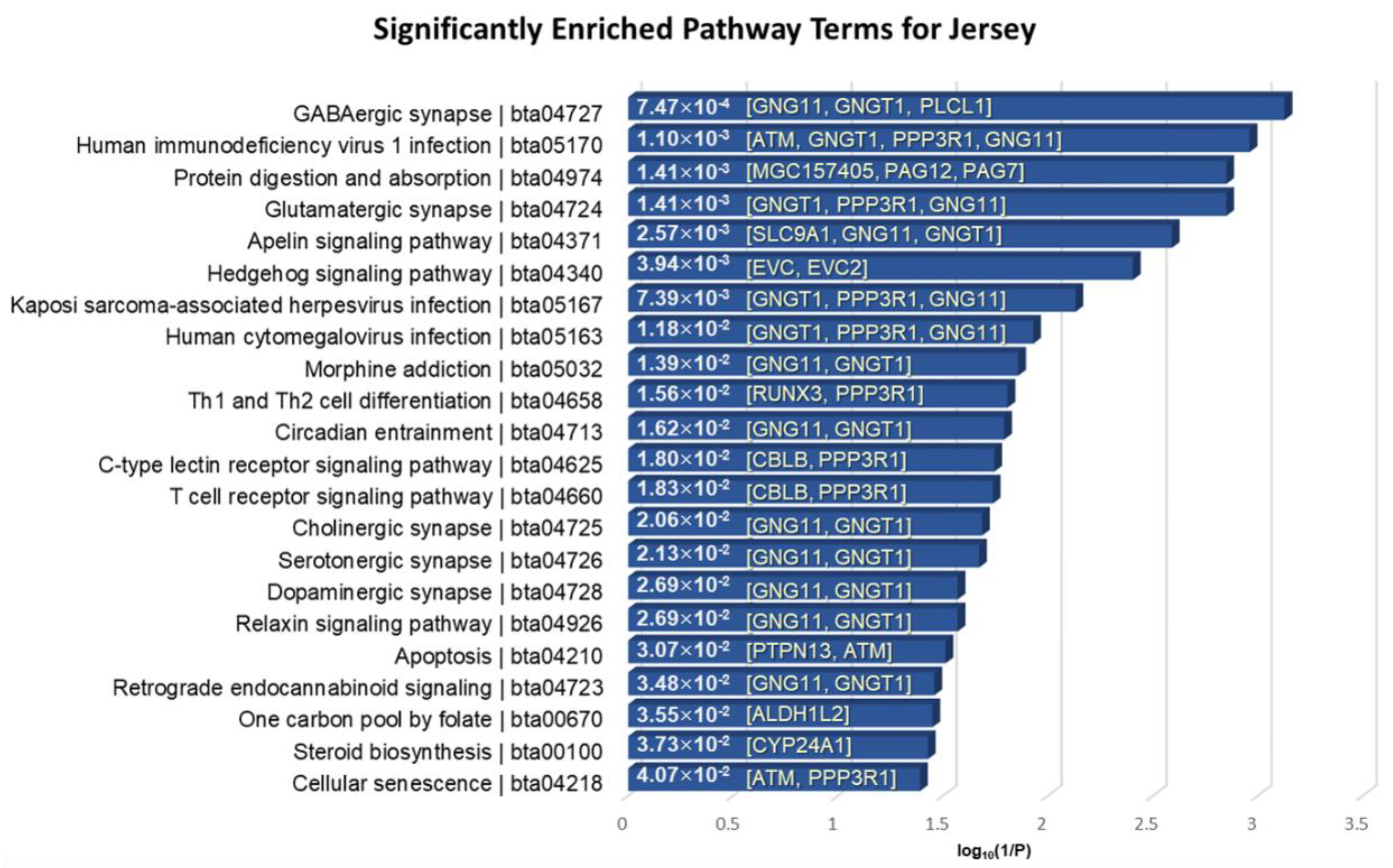
3.5. Functional Enrichment Analysis of the Identified Genes
3.6. Distinct Genetic Variation on the Mitochondrial Genome
3.7. Analysis of the Overlapped Genetic Signatures Using Diverse Statistics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ciepłoch, A.; Rutkowska, K.; Oprządek, J.; Poławska, E. Genetic disorders in beef cattle: A review. Genes Genom. 2017, 39, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Mai, M.D.; Sahana, G.; Christiansen, F.B.; Guldbrandtsen, B. A genome-wide association study for milk production traits in Danish Jersey cattle using a 50K single nucleotide polymorphism chip. J. Anim. Sci. 2010, 88, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Chamberlain, A.J.; Maceachern, S.; Savin, K.; McPartlan, H.; MacLeod, I.; Sethuraman, L.; Goddard, M.E. A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim. Genet. 2009, 40, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Rothammer, S.; Seichter, D.; Förster, M.; Medugorac, I. A genome-wide scan for signatures of differential artificial selection in ten cattle breeds. BMC Genom. 2013, 14, 908. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, Y.T.; Pérez O’Brien, A.M.; Sonstegard, T.S.; Van Tassell, C.P.; do Carmo, A.S.; Mészáros, G.; Sölkner, J.; Garcia, J.F. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS ONE 2013, 8, e64280. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, J.; Lee, T.; Son, J.K.; Yoon, H.B.; Baek, K.S.; Jeong, J.Y.; Cho, Y.M.; Lee, K.T.; Yang, B.C.; et al. Deciphering the genetic blueprint behind Holstein milk proteins and production. Genome Biol. Evol. 2014, 6, 1366–1374. [Google Scholar] [CrossRef]
- Taye, M.; Lee, W.; Jeon, S.; Yoon, J.; Dessie, T.; Hanotte, O.; Mwai, O.A.; Kemp, S.; Cho, S.; Oh, S.J.; et al. Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mamm. Genome 2017, 28, 528–541. [Google Scholar] [CrossRef]
- Xu, L.; Yang, L.; Zhu, B.; Zhang, W.; Wang, Z.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; et al. Genome-wide scan reveals genetic divergence and diverse adaptive selection in Chinese local cattle. BMC Genom. 2019, 20, 494. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Chen, H.; Patterson, N.; Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Shlyakhter, I.; Karlsson, E.K.; Byrne, E.H.; Morales, S.; Frieden, G.; Hostetter, E.; Angelino, E.; Garber, M.; Zuk, O.; et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 2010, 327, 883–886. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Nekrutenko, A.; Taylor, J. Next-generation sequencing data interpretation: Enhancing reproducibility and accessibility. Nat. Rev. Genet. 2012, 13, 667–672. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1897. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-encoding variational Bayes. In Proceedings of the Second International Conference on Learning Representations, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Bassily, R.; Nissim, K.; Smith, A.; Steinke, T.; Stemmer, U.; Ullman, J. Algorithmic stability for adaptive data analysis. In Proceedings of the Forty-Eighth Annual ACM Symposium on Theory of Computing, Cambridge, MA, USA, 19–21 June 2016; pp. 1046–1059. [Google Scholar]
- Na, Y.J.; Sohn, K.A.; Kim, J.H. Interpretation of personal genome sequencing data in terms of disease ranks based on mutual information. BMC Med. Genom. 2015, 8, S4. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.; Feltus, F.A.; Park, J.P.; Coissieux, M.M.; Chang, C.; Chan, V.B.S.; Bentires-Alj, M.; Booth, B.W. Cancer cell redirection biomarker discovery using a mutual information approach. PLoS ONE 2017, 12, e0179265. [Google Scholar] [CrossRef]
- Wyner, A.D. A definition of conditional mutual information for arbitrary ensembles. Inf. Control 1978, 38, 51–59. [Google Scholar] [CrossRef]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory, 2nd ed.; Wiley: New York, NY, USA, 2006. [Google Scholar]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Inouye, M. Fast principal component analysis of large-scale genome-wide data. PLoS ONE 2014, 9, e93766. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Coop, G.; Novembre, J.; Kudaravalli, S.; Li, J.Z.; Absher, D.; Srinivasan, B.S.; Barsh, G.; Myers, R.M.; Feldman, M.W.; et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009, 19, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Ogorevc, J.; Kunej, T.; Razpet, A.; Dovc, P. Database of cattle candidate genes and genetic markers for milk production and mastitis. Anim. Genet. 2009, 40, 832–851. [Google Scholar] [CrossRef]
- Ron, M.; Israeli, G.; Seroussi, E.; Weller, J.I.; Gregg, J.P.; Shani, M.; Medrano, J.F. Combining mouse mammary gland gene expression and comparative mapping for the identification of candidate genes for QTL of milk production traits in cattle. BMC Genom. 2007, 8, 183. [Google Scholar] [CrossRef]
- Clark, D.L.; Boler, D.D.; Kutzler, L.W.; Jones, K.A.; McKeith, F.K.; Killefer, J.; Carr, T.R.; Dilger, A.C. Muscle gene expression associated with increased marbling in beef cattle. Anim. Biotechnol. 2011, 22, 51–63. [Google Scholar] [CrossRef] [PubMed]
- De Jager, N.; Hudson, N.J.; Reverter, A.; Barnard, R.; Café, L.M.; Greenwood, P.L.; Dalrymple, B.P. Gene expression phenotypes for lipid metabolism and intramuscular fat in skeletal muscle of cattle. J. Anim. Sci. 2013, 91, 1112–1128. [Google Scholar] [CrossRef]
- Lim, D.; Kim, N.K.; Park, H.S.; Lee, S.H.; Cho, Y.M.; Oh, S.J.; Kim, T.H.; Kim, H. Identification of candidate genes related to bovine marbling using protein-protein interaction networks. Int. J. Biol. Sci. 2011, 7, 992–1002. [Google Scholar] [CrossRef]
- Wang, Y.H.; Bower, N.I.; Reverter, A.; Tan, S.H.; De Jager, N.; Wang, R.; McWilliam, S.M.; Café, L.M.; Greenwood, P.L.; Lehnert, S.A. Gene expression patterns during intramuscular fat development in cattle. J. Anim. Sci. 2009, 87, 119–130. [Google Scholar] [CrossRef]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef] [PubMed]
- Harvatine, K.J.; Perfield, J.W.; Bauman, D.E. Expression of enzymes and key regulators of lipid synthesis is upregulated in adipose tissue during CLA-induced milk fat depression in dairy cows. J. Nutr. 2009, 139, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kwon, E.G.; Im, S.K.; Seo, K.S.; Baik, M. Expression of fat deposition and fat removal genes is associated with intramuscular fat content in longissimus dorsi muscle of Korean cattle steers. J. Anim. Sci. 2012, 90, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Duckett, S.K.; Azain, M.J.; Sonon, R.N.; Pringle, T.D., Jr. The effect of anabolic implants on intramuscular lipid deposition in finished beef cattle. J. Anim. Sci. 2007, 85, 430–440. [Google Scholar] [CrossRef]
- Estany, J.; Ros-Freixedes, R.; Tor, M.; Pena, R.N. A functional variant in the stearoyl-coA desaturase gene promoter enhances fatty acid desaturation in pork. PLoS ONE 2014, 9, e86177. [Google Scholar] [CrossRef]
- Rincon, G.; Islas-Trejo, A.; Castillo, A.R.; Bauman, D.E.; German, B.J.; Medrano, J.F. Polymorphisms in genes in the SREBP1 signaling pathway and SCD are associated with milk fatty acid composition in Holstein cattle. J. Dairy Res. 2012, 79, 66–75. [Google Scholar] [CrossRef]
- Dong, X.Y.; Tang, S.Q. Insulin-induced gene: A new regulator in lipid metabolism. Peptides 2010, 31, 2145–2150. [Google Scholar] [CrossRef]
- Komisarek, J.; Michalak, A.; Walendowska, A. The effects of polymorphisms in DGAT1, GH and GHR genes on reproduction and production traits in Jersey cows. Anim. Sci. 2011, 29, 29–36. [Google Scholar]
- Yamada, T. Genetic dissection of marbling trait through integration of mapping and expression profiling. Anim. Sci. J. 2014, 85, 349–355. [Google Scholar] [CrossRef]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Weikard, R.; Kühn, C.; Goldammer, T.; Freyer, G.; Schwerin, M. The bovine PPARGC1A gene: Molecular characterization and association of an SNP with variation of milk fat synthesis. Physiol. Genom. 2005, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hutley, L.; Shurety, W.; Newell, F.; McGeary, R.; Pelton, N.; Grant, J.; Herington, A.; Cameron, D.; Whitehead, J.; Prins, J. Fibroblast growth factor 1: A key regulator of human adipogenesis. Diabetes 2004, 53, 3097–3106. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ahn, C.; Bong, N.; Choe, S.; Lee, D.K. Biphasic effects of FGF2 on adipogenesis. PLoS ONE 2015, 10, e0120073. [Google Scholar] [CrossRef] [PubMed]
- Holly, J.; Sabin, M.; Perks, C.; Shield, J. Adipogenesis and IGF-1. Metab. Syndr. Relat. Disord. 2006, 4, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Casas, E.; Keele, J.W.; Shackelford, S.D.; Koohmaraie, M.; Stone, R.T. Identification of quantitative trait loci for growth and carcass composition in cattle. Anim. Genet. 2004, 35, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Sasaki, S.; Sukegawa, S.; Yoshioka, S.; Takahagi, Y. Association of a single nucleotide polymorphism in titin gene with marbling in Japanese Black beef cattle. BMC Res. Notes 2009, 2, 78. [Google Scholar] [CrossRef]
- Kuceriva, J.; Matejicek, A.; Jandurova, O.M.; Sørensen, P.; Nemcova, E.; Stipkova, M.; Kott, T.; Bouska, J.; Frelich, J. Milk protein genes CSN1S1, CSN2, CSN3, LGB and their relation to genetic values of milk production parameters in Czech Fleckvieh. Czech J. Anim. Sci. 2006, 6, 241–247. [Google Scholar]
- Lemay, D.G.; Lynn, D.J.; Martin, W.F.; Neville, M.C.; Casey, T.M.; Rincon, G.; Kriventseva, E.V.; Barris, W.C.; Hinrichs, A.S.; Molenaar, A.J.; et al. The bovine lactation genome: Insights into the evolution of mammalian milk. Genome Biol. 2009, 10, R43. [Google Scholar] [CrossRef]
- Bonfatti, V.; Giantin, M.; Gervaso, M.; Coletta, A.; Dacasto, M.; Carnier, P. Effect of CSN1S1-CSN3 (α(S1)-κ-casein) composite genotype on milk production traits and milk coagulation properties in Mediterranean water buffalo. J. Dairy Sci. 2012, 95, 3435–3443. [Google Scholar] [CrossRef]
- Aoki, N.; Ishii, T.; Ohira, S.; Yamaguchi, Y.; Negi, M.; Adachi, T.; Nakamura, R.; Matsuda, T. Stage specific expression of milk fat globule membrane glycoproteins in mouse mammary gland: Comparison of MFG-E8, butyrophilin, and CD36 with a major milk protein, beta-casein. Biochim. Biophys. Acta 1997, 1334, 182–190. [Google Scholar] [CrossRef]
- Dowbenko, D.; Kikuta, A.; Fennie, C.; Gillett, N.; Lasky, L.A. Glycosylation-dependent cell adhesion molecule 1 (GlyCAM 1) mucin is expressed by lactating mammary gland epithelial cells and is present in milk. J. Clin. Investig. 1993, 92, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Georges, M. Domestic-animal genomics: Deciphering the genetics of complex traits. Nat. Rev. Genet. 2004, 5, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Hadjiconstantouras, C.; Sargent, C.A.; Skinner, T.M.; Archibald, A.L.; Haley, C.S.; Plastow, G.S. Characterization of the porcine KIT ligand gene: Expression analysis, genomic structure, polymorphism detection and association with coat colour traits. Anim. Genet. 2008, 39, 217–224. [Google Scholar] [CrossRef]
- Petit, R.J.; Mousadik, A.; Pons, O. Identifying populations for conservation on the basis of genetic markers. Cons. Biol. 1997, 12, 844–855. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Lim, J.H.; Song, M.J.; Kim, O.H.; Park, B.Y.; Kim, M.J.; Hwang, I.H.; Lee, S.C. Comparisons of longissimus muscle metabolic enzymes and muscle fiber types in Korean and western pig breeds. Meat Sci. 2008, 78, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Cho, Y.M.; Jung, Y.S.; Kim, G.S.; Heo, K.N.; Lee, S.H.; Lim, D.; Cho, S.; Park, E.W.; Yoon, D. Gene expression profiling of metabolism-related genes between top round and loin muscle of Korean cattle (Hanwoo). J. Agric. Food Chem. 2009, 57, 10898–10903. [Google Scholar] [CrossRef]
- Jurie, C.; Cassar-Malek, I.; Bonnet, M.; Leroux, C.; Bauchart, D.; Boulesteix, P.; Pethick, D.W.; Hocquette, J.F. Adipocyte fatty acid binding protein and mitochondrial enzyme activities in muscles as relevant indicators of marbling in cattle. J. Anim. Sci. 2007, 85, 2660–2669. [Google Scholar] [CrossRef]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef]
- Schleinitz, D.; Klöting, N.; Lindgren, C.M.; Breitfeld, J.; Dietrich, A.; Schön, M.R.; Lohmann, T.; Dreßler, M.; Stumvoll, M.; McCarthy, M.I.; et al. Fat depot-specific mRNA expression of novel loci associated with waist-hip ratio. Int. J. Obes. 2014, 38, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, Y.; Wang, G.; Li, H. Identification of microRNA and bioinformatics target gene analysis in beef cattle intramuscular fat and subcutaneous fat. Mol. Biosyst. 2013, 9, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.; Yoon, J.; Dessie, T.; Cho, S.; Oh, S.J.; Lee, H.K.; Kim, H. Deciphering signature of selection affecting beef quality traits in Angus cattle. Genes Genom. 2018, 40, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Wang, F.; Liu, C.; Xu, X.; Liu, B. TEAD1-dependent expression of the FoxO3a gene in mouse skeletal muscle. BMC Mol. Biol. 2011, 12, 1. [Google Scholar] [CrossRef]
- Wijga, S.; Bastiaansen, J.W.; Wall, E.; Strandberg, E.; de Haas, Y.; Giblin, L.; Bovenhuis, H. Genomic associations with somatic cell score in first-lactation Holstein cows. J. Dairy Sci. 2012, 95, 899–908. [Google Scholar] [CrossRef]
- Raven, L.A.; Cocks, B.G.; Hayes, B.J. Multibreed genome wide association can improve precision of mapping causative variants underlying milk production in dairy cattle. BMC Genom. 2014, 15, 62. [Google Scholar] [CrossRef]
- Shin, D.; Lee, C.; Park, K.D.; Kim, H.; Cho, K.H. Genome-association analysis of Korean Holstein milk traits using genomic estimated breeding value. Asian-Australas. J. Anim. Sci. 2017, 30, 309–319. [Google Scholar]
- Patton, S.; Kesler, E.M. Saturation in milk and meat fats. Science 1967, 156, 1365–1366. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Shadyab, A.H.; LaCroix, A.Z. Genetic factors associated with longevity: A review of recent findings. Ageing Res. Rev. 2015, 19, 1–7. [Google Scholar] [CrossRef]
- Fernández, A.I.; Alves, E.; Fernández, A.; de Pedro, E.; López-García, M.A.; Ovilo, C.; Rodriguez, M.C.; Silio, L. Mitochondrial genome polymorphisms associated with longissimus muscle composition in Iberian pigs. J. Anim. Sci. 2008, 86, 1283–1290. [Google Scholar]
- Wang, J.; Xiang, H.; Liu, L.; Kong, M.; Yin, T.; Zhao, X. Mitochondrial haplotypes influence metabolic traits across bovine inter- and intra-species cybrids. Sci. Rep. 2017, 7, 4179. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.W.; Laipis, P.J. Mitochondrial DNA polymorphism in maternal lineages of Holstein cows. Proc. Nat. Acad. Sci. USA 1982, 79, 4686–4690. [Google Scholar] [CrossRef] [PubMed]
- Sutarno; Cummins, J.M.; Greeff, J.; Lymbery, A.J. Mitochondrial DNA polymorphisms and fertility in beef cattle. Theriogenology 2002, 57, 1603–1610. [Google Scholar] [CrossRef]
- Ron, M.; Genis, I.; Ezra, E.; Yoffe, O.; Weller, J.I.; Shani, M. Mitochondrial DNA polymorphism and determination of effects on economic traits in dairy cattle. Anim. Biotechnol. 1992, 3, 201–219. [Google Scholar] [CrossRef]
- Schutza, M.M.; Freeman, A.E.; Lindberg, G.L.; Koehler, C.M.; Beitz, D.C. The effect of mitochondrial DNA on milk production and health of dairy cattle. Livest. Prod. Sci. 1994, 37, 283–295. [Google Scholar] [CrossRef]
- Oleksyk, T.K.; Smith, M.W.; O’Brien, S.J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 185–205. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average of Heterozygosity Frequency of Lipid-Related Genes in Angus | |
| Total heterozygosity frequency in Angus | 0.391 |
| With homozygosity in Jersey | 2.813 |
| With heterozygosity in Jersey | 0 |
| Average of heterozygosity frequency of milk-related genes in Jersey | |
| Total heterozygosity frequency in Jersey | 0.431 |
| With homozygosity in Angus | 4.046 |
| With heterozygosity in Angus | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-J.; Ha, J.-W.; Kim, H. Genome-Wide Identification of Discriminative Genetic Variations in Beef and Dairy Cattle via an Information-Theoretic Approach. Genes 2020, 11, 678. https://doi.org/10.3390/genes11060678
Kim S-J, Ha J-W, Kim H. Genome-Wide Identification of Discriminative Genetic Variations in Beef and Dairy Cattle via an Information-Theoretic Approach. Genes. 2020; 11(6):678. https://doi.org/10.3390/genes11060678
Chicago/Turabian StyleKim, Soo-Jin, Jung-Woo Ha, and Heebal Kim. 2020. "Genome-Wide Identification of Discriminative Genetic Variations in Beef and Dairy Cattle via an Information-Theoretic Approach" Genes 11, no. 6: 678. https://doi.org/10.3390/genes11060678
APA StyleKim, S.-J., Ha, J.-W., & Kim, H. (2020). Genome-Wide Identification of Discriminative Genetic Variations in Beef and Dairy Cattle via an Information-Theoretic Approach. Genes, 11(6), 678. https://doi.org/10.3390/genes11060678