Transcriptome Analysis of Skeletal Muscle Reveals Altered Proteolytic and Neuromuscular Junction Associated Gene Expressions in a Mouse Model of Cerebral Ischemic Stroke

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. MCAO Surgery and Tissue Collection
2.3. TTC Staining and Assessment of Infarct Volume
2.4. Assessment of Functional Outcomes
2.5. RNA Sequencing and Data Analysis
2.6. GO and KEGG Analyses
2.7. Detailed Functional Analysis via PubMed Search Terms
2.8. Real-Time PCR
3. Results
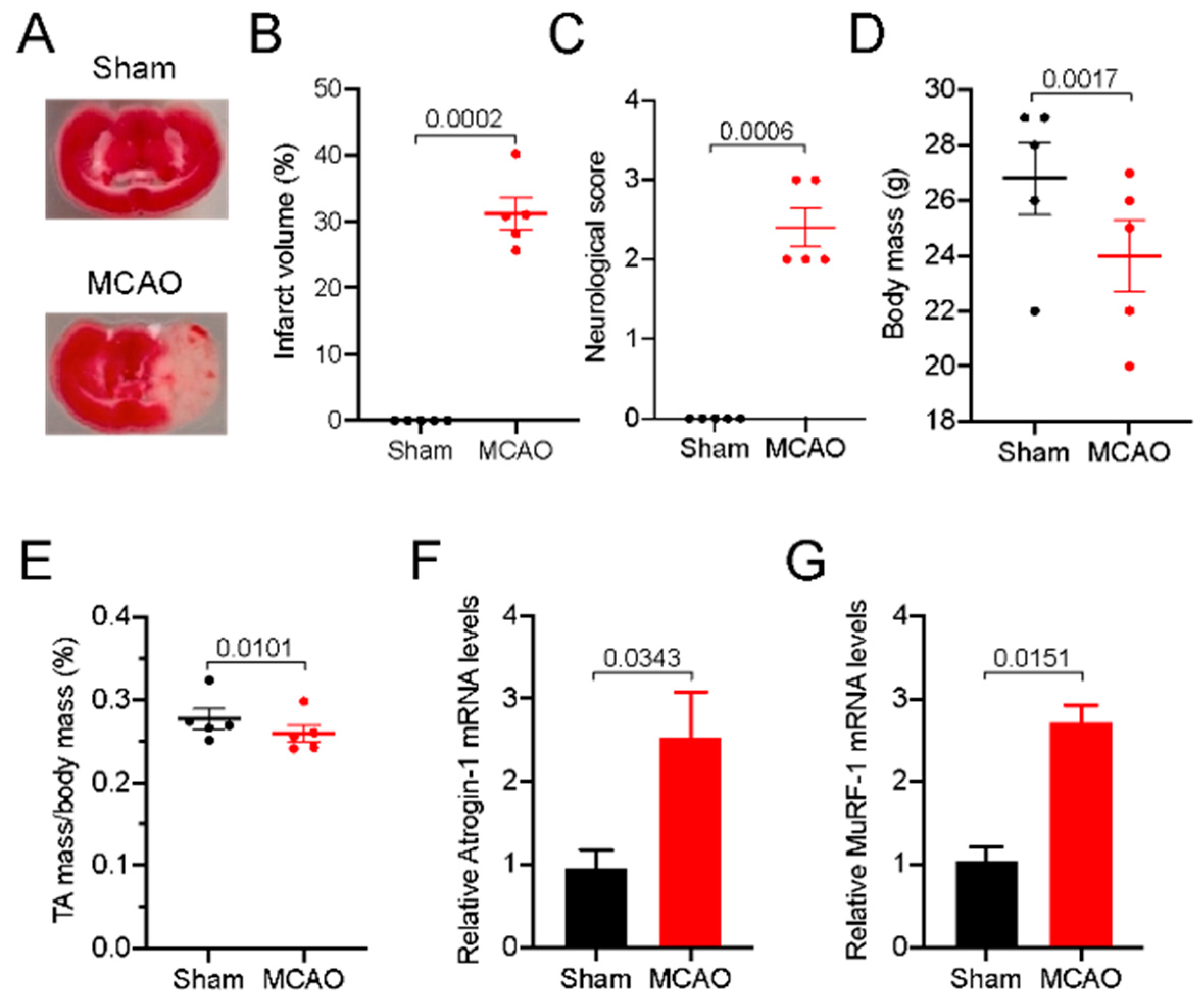
3.1. Stroke Robustly Induced Skeletal Muscle Atrophy
3.2. Stroke Significantly Altered Skeletal Muscle Transcriptome Profile
3.3. Profiling of Differentially Expressed Genes/Transcripts of Post-stroke Skeletal Muscle
3.4. GO Enrichment Analysis of Differentially Expressed Genes of Post-stroke Muscle
3.5. KEGG Enrichment Analysis of Differentially Expressed Genes
3.6. Stroke Induced Differential Expression of Neuromuscular Junction-Associated Genes
3.7. Differentially Expressed Ubiquitin–Proteasome Genes in Post-Stroke Muscle
3.8. Stroke altered the Genes Associated with PI3K-Akt-mTOR Pathway
3.9. Stroke Altered p53 Pathway- and Cell-Cycle-Associated Genes
3.10. Numerous Extracellular Matrix Associated Genes are Disturbed in Post-stroke Muscle
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Feigin, V.L.; Roth, G.A.; Naghavi, M.; Parmar, P.; Krishnamurthi, R.; Chugh, S.; Mensah, G.A.; Norrving, B.; Shiue, I.; Ng, M. Global burden of stroke and risk factors in 188 countries, during 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016, 15, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Scherbakov, N.; Von Haehling, S.; Anker, S.D.; Dirnagl, U.; Doehner, W. Stroke induced Sarcopenia: Muscle wasting and disability after stroke. Int. J. Cardiol. 2013, 170, 89–94. [Google Scholar] [CrossRef]
- English, C.; McLennan, H.; Thoirs, K.; Coates, A.; Bernhardt, J. Loss of skeletal muscle mass after stroke: A systematic review. Int. J. Stroke 2010, 5, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Dobkin, B.H. Training and exercise to drive poststroke recovery. Nat. Clin. Pract. Neurol. 2008, 4, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Saunders, D.H.; Greig, C.A.; Young, A.; Mead, G.E. Physical fitness training for stroke patients. Stroke 2004, 35, 2235. [Google Scholar] [CrossRef] [Green Version]
- Dobkin, B.H. Clinical practice. Rehabilitation after stroke. N. Engl. J. Med. 2005, 352, 1677–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Francisco, G.E. New insights into the pathophysiology of post-stroke spasticity. Front. Hum. Neurosci. 2015, 9, 192. [Google Scholar] [CrossRef] [Green Version]
- Bouziana, S.D.; Tziomalos, K. Malnutrition in patients with acute stroke. J. Nutr. Metab. 2011, 45, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Anrather, J.; Iadecola, C. Inflammation and stroke: An overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef]
- Desgeorges, M.M.; Devillard, X.; Toutain, J.; Divoux, D.; Castells, J.; Bernaudin, M.; Touzani, O.; Freyssenet, D.G. Molecular mechanisms of skeletal muscle atrophy in a mouse model of cerebral ischemia. Stroke 2015, 46, 1673–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desgeorges, M.M.; Devillard, X.; Toutain, J.; Castells, J.; Divoux, D.; Arnould, D.F.; Haqq, C.; Bernaudin, M.; Durieux, A.-C.; Touzani, O.; et al. Pharmacological inhibition of myostatin improves skeletal muscle mass and function in a mouse model of stroke. Sci. Rep. UK 2017, 7, 14000. [Google Scholar] [CrossRef] [PubMed]
- Freire, P.P.; Fernandez, G.J.; Cury, S.S.; de Moraes, D.; Oliveira, J.S.; de Oliveira, G.; Dal-Pai-Silva, M.; dos Reis, P.P.; Carvalho, R.F. The Pathway to Cancer Cachexia: MicroRNA-Regulated Networks in Muscle Wasting Based on Integrative Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llano-Diez, M.; Fury, W.; Okamoto, H.; Bai, Y.; Gromada, J.; Larsson, L. RNA-sequencing reveals altered skeletal muscle contraction, E3 ligases, autophagy, apoptosis, and chaperone expression in patients with critical illness myopathy. Skelet. Muscle 2019, 9, 9. [Google Scholar] [CrossRef]
- Khan, M.M.; Gandhi, C.; Chauhan, N.; Stevens, J.W.; Motto, D.G.; Lentz, S.R.; Chauhan, A.K. Alternatively-Spliced Extra Domain a of Fibronectin Promotes Acute Inflammation and Brain Injury After Cerebral Ischemia in Mice. Stroke 2012, 43, 1376. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.M.; Motto, D.G.; Lentz, S.R.; Chauhan, A.K. ADAMTS13 reduces VWF-mediated acute inflammation following focal cerebral ischemia in mice. J. Thromb. Haemost 2012, 10, 1665–1671. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.A.; Morton, M.T.; Tsao-Wu, G.; Savalos, R.A.; Davidson, C.; Sharp, F.R. A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 1990, 10, 290–293. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [Green Version]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, J.S.; Lopez, M.A.; Boriek, A.M. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3beta. J. Biol. Chem. 2010, 285, 29336–29347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, J.S.; Lopez, M.A.; Cox, G.A.; Boriek, A.M. Anisotropic regulation of Ankrd2 gene expression in skeletal muscle by mechanical stretch. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3330–3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesquita, C. Semantic Similarity in the Gene Ontology. In The Gene Ontology Handbook; Dessimoz, C., Škunca, N., Eds.; Springer: New York, NY, USA, 2017; pp. 161–173. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, J.; Rajapakse, J.C. Gene Ontology Enrichment Improves Performances of Functional Similarity of Genes. Sci. Rep. 2018, 8, 12100. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Sarkar, R.R. Comparison of human cell signaling pathway databases—Evolution, drawbacks and challenges. Database 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Dickson, H.M.; Wilbur, A.; Reinke, A.A.; Young, M.A.; Vojtek, A.B. Targeted inhibition of the Shroom3-Rho kinase protein-protein interaction circumvents Nogo66 to promote axon outgrowth. BMC Neurosci. 2015, 16, 34. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R. MuSK function during health and disease. Neurosci. Lett. 2020, 716, 134676. [Google Scholar] [CrossRef]
- Burden, S.J.; Yumoto, N.; Zhang, W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a009167. [Google Scholar] [CrossRef] [Green Version]
- von Grabowiecki, Y.; Abreu, P.; Blanchard, O.; Palamiuc, L.; Benosman, S.; Meriaux, S.; Devignot, V.; Gross, I.; Mellitzer, G.; de Aguilar, J.L.G. Transcriptional activator TAp63 is upregulated in muscular atrophy during ALS and induces the pro-atrophic ubiquitin ligase Trim63. Elife 2016, 5, e10528. [Google Scholar] [CrossRef]
- Macpherson, P.C.D.; Wang, X.; Goldman, D. Myogenin regulates denervation-dependent muscle atrophy in mouse soleus muscle. J. Cell Biochem. 2011, 112, 2149–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, N.; Hirasaka, K.; Maeda, T.; Haruna, M.; Shiota, C.; Ochi, A.; Abe, T.; Kohno, S.; Ohno, A.; Teshima-Kondo, S. Prevention of skeletal muscle atrophy in vitro using anti-ubiquitination oligopeptide carried by atelocollagen. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 873–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagiya, A.; Suyama, E.; Adachi, H.; Svitkin, Y.V.; Aza-Blanc, P.; Imataka, H.; Mikami, S.; Martineau, Y.; Ronai, Z.e.A.; Sonenberg, N. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol. Cell 2012, 46, 847–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.-I.; Katz, M.; Chin, S.; Qi, X.; Cruz, J.; Ibebunjo, C.; Zhao, S.; Chen, A.; Glass, D.J. MNK2 Inhibits eIF4G Activation Through a Pathway Involving Serine-Arginine–Rich Protein Kinase in Skeletal Muscle. Sci. Signal. 2012, 5, ra14. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.M.; Dyle, M.C.; Kunkel, S.D.; Bullard, S.A.; Bongers, K.S.; Fox, D.K.; Dierdorff, J.M.; Foster, E.D.; Adams, C.M. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem. 2012, 287, 27290–27301. [Google Scholar] [CrossRef] [Green Version]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21CDKN1A in the DNA damage response. Mutat. Res. Rev. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef]
- Kong, D.; He, M.; Yang, L.; Zhou, R.; Yan, Y.-Q.; Liang, Y.; Teng, C.-B. MiR-17 and miR-19 cooperatively promote skeletal muscle cell differentiation. Cell. Mol. Life Sci. 2019, 76, 5041–5054. [Google Scholar] [CrossRef] [Green Version]
- Segalés, J.; Perdiguero, E.; Serrano, A.L.; Sousa-Victor, P.; Ortet, L.; Jardí, M.; Budanov, A.V.; Garcia-Prat, L.; Sandri, M.; Thomson, D.M. Sestrin prevents atrophy of disused and aging muscles by integrating anabolic and catabolic signals. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Fernando, P.; Kelly, J.F.; Balazsi, K.; Slack, R.S.; Megeney, L.A. Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 11025–11030. [Google Scholar] [CrossRef] [Green Version]
- Larsen, B.D.; Rampalli, S.; Burns, L.E.; Brunette, S.; Dilworth, F.J.; Megeney, L.A. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 4230–4235. [Google Scholar] [CrossRef] [Green Version]
- Hunnicutt, J.L.; Gregory, C.M. Skeletal muscle changes following stroke: A systematic review and comparison to healthy individuals. Top. Stroke Rehabil. 2017, 24, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.; Messing, R.O.; Chou, W.-H. Mouse model of middle cerebral artery occlusion. J. Vis. Exp. JoVE 2011, 2761. [Google Scholar] [CrossRef] [Green Version]
- Yampolsky, P.; Pacifici, P.G.; Witzemann, V. Differential muscle-driven synaptic remodeling in the neuromuscular junction after denervation. Eur. J. Neurosci. 2010, 31, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Yee, W.C.; Pestronk, A. Mechanisms of postsynaptic plasticity: Remodeling of the junctional acetylcholine receptor cluster induced by motor nerve terminal outgrowth. J. Neurosci. 1987, 7, 2019–2024. [Google Scholar] [CrossRef] [Green Version]
- Castets, P.; Rion, N.; Théodore, M.; Falcetta, D.; Lin, S.; Reischl, M.; Wild, F.; Guérard, L.; Eickhorst, C.; Brockhoff, M. mTORC1 and PKB/Akt control the muscle response to denervation by regulating autophagy and HDAC4. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, J.E.; Bergqvist, C.A.; Larhammar, D. Evolution of vertebrate nicotinic acetylcholine receptors. BMC Evol. Biol. 2019, 19, 38. [Google Scholar] [CrossRef] [Green Version]
- Hallock, P.T.; Chin, S.; Blais, S.; Neubert, T.A.; Glass, D.J. Sorbs1 and -2 Interact with CrkL and Are Required for Acetylcholine Receptor Cluster Formation. Mol. Cell. Biol. 2015, 36, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ip, F.C.; Shi, L.; Zhang, Z.; Tang, H.; Ng, Y.P.; Ye, W.-C.; Fu, A.K.; Ip, N.Y. Coronin 6 regulates acetylcholine receptor clustering through modulating receptor anchorage to actin cytoskeleton. J. Neurosci. 2014, 34, 2413–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.C.; Barzaghi, P.; Ruegg, M.A. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep. 2004, 5, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesser, B.A.; Henschel, O.; Witzemann, V. Synapse disassembly and formation of new synapses in postnatal muscle upon conditional inactivation of MuSK. Mol. Cell. Neurosci. 2006, 31, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esumi, E.; Li, S.; Nonaka, I.; Nabeshima, Y. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Siu, P.M.; Donley, D.A.; Bryner, R.W.; Alway, S.E. Myogenin and oxidative enzyme gene expression levels are elevated in rat soleus muscles after endurance training. J. Appl. Physiol. 2004, 97, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Tsai, L.K.; Liao, K.Y.; Wu, T.C.; Huang, Y.H.; Huang, Y.C.; Chang, S.W.; Wang, P.Y.; Tsao, Y.P.; Chen, S.L. Muscle-restricted nuclear receptor interaction protein knockout causes motor neuron degeneration through down-regulation of myogenin at the neuromuscular junction. J. Cachexia Sarcopenia Muscle 2018, 9, 771–785. [Google Scholar] [CrossRef]
- Furuya, N.; Ikeda, S.-I.; Sato, S.; Soma, S.; Ezaki, J.; Trejo, J.A.O.; Takeda-Ezaki, M.; Fujimura, T.; Arikawa-Hirasawa, E.; Tada, N. PARK2/Parkin-mediated mitochondrial clearance contributes to proteasome activation during slow-twitch muscle atrophy via NFE2L1 nuclear translocation. Autophagy 2014, 10, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.C.; Cheng, Z.; Copps, K.D.; White, M.F. Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol. Cell. Biol. 2011, 31, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Sarah Eckstein, S.; Weigert, C.; Lehmann, R. Divergent roles of IRS (insulin receptor substrate) 1 and 2 in liver and skeletal muscle. Curr. Med. Chem. 2017, 24, 1827–1852. [Google Scholar] [CrossRef]
- Lee, D.; Takayama, S.; Goldberg, A.L. ZFAND5/ZNF216 is an activator of the 26S proteasome that stimulates overall protein degradation. Proc. Natl. Acad. Sci. USA 2018, 115, E9550–E9559. [Google Scholar] [CrossRef] [Green Version]
- Heng, A.; Ventadour, S.; Jarzaguet, M.; Pouch-Pelissier, M.; Guezennec, C.; Bigard, X.; Attaix, D.; Taillandier, D. Coordinate expression of the 19S regulatory complex and evidence for ubiquitin-dependent telethonin degradation in the unloaded soleus muscle. Int. J. Biochem. Cell Biol. 2008, 40, 2544–2552. [Google Scholar] [CrossRef]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef]
- Song, Y.-H.; Li, Y.; Du, J.; Mitch, W.E.; Rosenthal, N.; Delafontaine, P. Muscle-specific expression of IGF-1 blocks angiotensin II–induced skeletal muscle wasting. J. Clin. Investig. 2005, 115, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llano-Diez, M.; Gustafson, A.-M.; Olsson, C.; Goransson, H.; Larsson, L. Muscle wasting and the temporal gene expression pattern in a novel rat intensive care unit model. BMC Genom. 2011, 12, 602. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Qian, L.; Zhang, J.; Zhang, W.; Morrison, A.; Hayes, P.; Wilson, S.; Chen, T.; Zhao, J. Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J. Neurosci. Res. 2011, 89, 1723–1736. [Google Scholar] [CrossRef]
- Bullard, S.A.; Seo, S.; Schilling, B.; Dyle, M.C.; Dierdorff, J.M.; Ebert, S.M.; DeLau, A.D.; Gibson, B.W.; Adams, C.M. Gadd45a Protein Promotes Skeletal Muscle Atrophy by Forming a Complex with the Protein Kinase MEKK4. J. Biol. Chem. 2016, 291, 17496–17509. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, C.; Svensson, A.; Christerson, U.; Tågerud, S. Denervation-induced alterations in gene expression in mouse skeletal muscle. Eur. J. Neurosci. 2005, 21, 577–580. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y. Role of matrix metalloproteinases in skeletal muscle: Migration, differentiation, regeneration and fibrosis. Cell Adh. Migr. 2009, 3, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Yeghiazaryan, M.; Żybura-Broda, K.; Cabaj, A.; Włodarczyk, J.; Sławińska, U.; Rylski, M.; Wilczyński, G.M. Fine-structural distribution of MMP-2 and MMP-9 activities in the rat skeletal muscle upon training: A study by high-resolution in situ zymography. Histochem. Cell Biol. 2012, 138, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SH1 | SH2 | SH3 | ST2 | ST3 | ST4 |
|---|---|---|---|---|---|---|
| Raw Data | ||||||
| Read | 94,947,264 | 93,442,210 | 97,166,968 | 82,470,476 | 68,558,874 | 74,086,190 |
| Base | 14.24G | 14.02G | 14.58G | 12.37G | 10.28G | 11.11G |
| Valid Data | ||||||
| Read | 49,031,300 | 45,492,816 | 43,434,064 | 49,971,332 | 39,457,490 | 44,043,614 |
| Base | 7.35G | 6.82G | 6.52G | 7.50G | 5.92G | 6.61G |
| Valid Ratio(reads) | 51.64 | 48.69 | 44.70 | 60.59 | 57.55 | 59.45 |
| Q20% | 99.98 | 99.98 | 99.98 | 99.98 | 99.98 | 99.98 |
| Q30% | 97.89 | 97.82 | 97.77 | 97.71 | 97.38 | 97.79 |
| GC content% | 50 | 49.50 | 49.50 | 50 | 50 | 49.50 |
| Mapped reads (%) | 47,311,899 (96.5) | 43,962,039 (96.6) | 41,872,802 (96.0) | 48,017,122 (96.1) | 37,866,250 (95.6) | 42,454,571 (96.4) |
| Unique Mapped reads (%) | 40,092,540 (81.8) | 37,989,504 (83.5) | 35,870,140 (82.6) | 41,470,662 (83) | 32,052,577 (81.2) | 36,442,450 (82.7) |
| Multi Mapped reads (%) | 7,219,359 (14.7) | 5,972,535 (13.1) | 6,002,662 (13.8) | 6,546,460 (13.1) | 5,813,673 (14.7) | 6,012,121 (13.6) |
| PE Mapped reads (%) | 42,315,514 (86.3) | 38,786,818 (85.3) | 35,419,862 (81.5) | 41,386,258 (82.8) | 34,974,768 (88.6) | 39,438,248 (89.5) |
| Reads map to sense strand (%) | 22,821,235 (46.5) | 21,287,151 (46.8) | 20,174,801 (46.4) | 23,269,087 (46.6) | 18,351,916 (46.5) | 20,683,838 (47) |
| Reads map to antisense strand (%) | 22,835,667 (46.6) | 21,311,300 (46.8) | 20,199,286 (46.5) | 23,293,772 (46.6) | 18,350,598 (46.5) | 20,686,564 (47.0) |
| Non-splice reads v | 29,191,582 (59.5) | 28,846,354 (63.4) | 26,973,027 (62.1) | 31,573,393 (63.1) | 23,944,091 (60.7) | 27,383,178 (62.1) |
| Splice reads (%) | 16,465,320 (33.6) | 13,752,097 (30.2) | 13,401,060 (30.8) | 14,989,466 (30.0) | 12,758,423 (32.0) | 13,987,224 (31.8) |
| Exon | 84.60 | 81.09 | 81.79 | 81.17 | 83.94 | 82.53 |
| Intron | 14.62 | 17.96 | 17.34 | 17.90 | 15.12 | 16.55 |
| Intergenic | 0.78 | 0.95 | 0.87 | 0.93 | 0.94 | 0.92 |
| Sample | SH1 | SH2 | SH3 | ST2 | ST3 | ST4 |
|---|---|---|---|---|---|---|
| Statistics of gene expression | ||||||
| Exp gene | 36,375 | 35,766 | 35,988 | 36,185 | 33,129 | 35,113 |
| Min. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 1st Qu. | 0.23 | 0.25 | 0.27 | 0.24 | 0.20 | 0.22 |
| Median | 0.71 | 0.80 | 0.92 | 0.77 | 0.66 | 0.70 |
| Mean | 6.90 | 6.24 | 7.15 | 6.17 | 6.16 | 5.51 |
| 3rd Qu. | 1.89 | 1.96 | 2.04 | 1.91 | 1.88 | 1.80 |
| Max. | 26,401.93 | 21,260.97 | 34,914.37 | 21,795.19 | 16,188.98 | 13,581.60 |
| Sd. | 191.68 | 147.86 | 227.22 | 151.37 | 122.48 | 108.64 |
| Sum. | 250,873.79 | 223,333.19 | 257,474.35 | 223,088.24 | 203,986.51 | 193,458.60 |
| Statistics of transcript expression | ||||||
| Exp transcripts | 86,885 | 84,306 | 83,971 | 84,811 | 77,785 | 82,780 |
| Min. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 1st Qu. | 0.15 | 0.16 | 0.17 | 0.15 | 0.14 | 0.14 |
| Median | 0.46 | 0.53 | 0.55 | 0.50 | 0.46 | 0.46 |
| Mean | 4.30 | 4.06 | 4.57 | 4.00 | 4.11 | 3.61 |
| 3rd Qu. | 1.26 | 1.43 | 1.46 | 1.38 | 1.36 | 1.29 |
| Max. | 26,577.91 | 21,402.67 | 35,147.07 | 21,940.43 | 16,296.84 | 13,672.09 |
| Sd. | 133.78 | 102.12 | 159.33 | 104.79 | 86.48 | 76.47 |
| Sum. | 373,865.71 | 342,630.21 | 383,566.99 | 339,116.11 | 319,482.85 | 298,438.48 |
| FPKM Interval (FI) of gene expression | ||||||
| 0–0.1 FI | 4881 (13.42%) | 4719 (13.19%) | 4386 (12.19%) | 4955 (13.69%) | 4913 (14.83%) | 4932 (14.05%) |
| 0.1–0.3 FI | 5956 (16.37%) | 5445 (15.22%) | 5231 (14.54%) | 5601 (15.48%) | 5999 (18.11%) | 5877 (16.74%) |
| 0.3–3.57 FI | 19,698 (54.15%) | 19,741 (55.19%) | 20,564 (57.14%) | 19,884 (54.95%) | 16,796 (50.70%) | 18,933 (53.92%) |
| 3.57–15 FI | 4306 (11.84%) | 4255 (11.90%) | 4303 (11.96%) | 4152 (11.47%) | 3811 (11.50%) | 3918 (11.16%) |
| 15–60 FI | 1181 (3.25%) | 1254 (3.51%) | 1162 (3.23%) | 1244 (3.44%) | 1246 (3.76%) | 1119 (3.19%) |
| >60 FI | 353 (0.97%) | 352 (0.98%) | 342 (0.95%) | 349 (0.96%) | 364 (1.10%) | 334 (0.95%) |
| FPKM Interval (FI) of transcripts expression | ||||||
| 0–0.1 FI | 16,742 (19.27%) | 15,316 (18.17%) | 14,704 (17.51%) | 15,921 (18.77%) | 15,449 (19.86%) | 16,322 (19.72%) |
| 0.1–0.3 FI | 17,136 (19.72%) | 15,413 (18.28%) | 15,320 (18.24%) | 16,014 (18.88%) | 15,489 (19.91%) | 16,644 (20.11%) |
| 0.3–3.57 FI | 44,076 (50.73%) | 44,123 (52.34%) | 44,891 (53.46%) | 43,671 (51.49%) | 37,956 (48.80%) | 41,312 (49.91%) |
| 3.57–15 FI | 6552 (7.54%) | 6986 (8.29%) | 6703 (7.98%) | 6783 (8.00%) | 6415 (8.25%) | 6225 (7.52%) |
| 15–60 FI | 1814 (2.09%) | 1888 (2.24%) | 1784 (2.12%) | 1866 (2.20%) | 1894 (2.43%) | 1740 (2.10%) |
| >60 FI | 565 (0.65%) | 580 (0.69%) | 569 (0.68%) | 556 (0.66%) | 582 (0.75%) | 537 (0.65%) |
| Transcript expression coverage | ||||||
| 0–1 | 29.61% | 29.87% | 30.08% | 29.25% | 34.54% | 32.32% |
| 2–5 | 39.13% | 38.06% | 37.86% | 37.82% | 36.91% | 38.56% |
| 6–10 | 14.12% | 14.99% | 15.89% | 15.08% | 12.58% | 13.45% |
| 11–15 | 4.80% | 4.83% | 4.93% | 5.16% | 4.43% | 4.44% |
| 16–20 | 2.66% | 2.73% | 2.58% | 2.75% | 2.36% | 2.40% |
| 21–25 | 1.71% | 1.74% | 1.64% | 1.75% | 1.62% | 1.54% |
| 26–30 | 1.18% | 1.16% | 1.08% | 1.27% | 1.16% | 1.04% |
| >30 | 6.79% | 6.62% | 5.94% | 6.92% | 6.40% | 6.25% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrandi, P.J.; Khan, M.M.; Paez, H.G.; Pitzer, C.R.; Alway, S.E.; Mohamed, J.S. Transcriptome Analysis of Skeletal Muscle Reveals Altered Proteolytic and Neuromuscular Junction Associated Gene Expressions in a Mouse Model of Cerebral Ischemic Stroke. Genes 2020, 11, 726. https://doi.org/10.3390/genes11070726
Ferrandi PJ, Khan MM, Paez HG, Pitzer CR, Alway SE, Mohamed JS. Transcriptome Analysis of Skeletal Muscle Reveals Altered Proteolytic and Neuromuscular Junction Associated Gene Expressions in a Mouse Model of Cerebral Ischemic Stroke. Genes. 2020; 11(7):726. https://doi.org/10.3390/genes11070726
Chicago/Turabian StyleFerrandi, Peter J., Mohammad Moshahid Khan, Hector G. Paez, Christopher R. Pitzer, Stephen E. Alway, and Junaith S. Mohamed. 2020. "Transcriptome Analysis of Skeletal Muscle Reveals Altered Proteolytic and Neuromuscular Junction Associated Gene Expressions in a Mouse Model of Cerebral Ischemic Stroke" Genes 11, no. 7: 726. https://doi.org/10.3390/genes11070726