Protective Mechanisms Against DNA Replication Stress in the Nervous System



Abstract
:1. Relevance of Genomic Stability for the Nervous System
2. Overview of DNA Replication Stress: How Cells Prevent and Respond
3. Cell Proliferation in Nervous System Development, Homeostasis, and Diseases
4. Replication Stress-Causing Factors in the Nervous System
4.1. DNA Polymerases
4.2. RecQ Family of DNA Helicases
4.2.1. WRN
4.2.2. BLM
4.2.3. RECQL4
4.3. Senataxin, Spinocerebellar Ataxia with Axonal Neuropathy 2, and Amyotrophic Lateral Sclerosis 4
4.4. Aicardi-Goutières’ Syndrome-Causing Genes
4.4.1. TREX1 Exonuclease
4.4.2. SAMHD1
4.4.3. RNAse H Ribonucleases and RNA Deaminase (ADAR1)
4.5. CTC1 and Telomere Maintenance
4.6. Fanconi Anemia-Causing Genes
4.7. XRCC1 and DNA Single-Strand Break Repair
5. Replication Stress Response in the Nervous System
5.1. PI-3 Kinases: ATR, ATM, and DNA-PK
5.1.1. ATR, ATRIP, and Seckel Syndrome
5.1.2. ATM
5.1.3. DNA-PK
5.2. The MRN Complex: Mre11, Rad50, and Nbs1
5.2.1. Ataxia-Telangiectasia-Like Disorder 1 (ATLD1)
5.2.2. Nijmegen Breakage Syndrome (NBS)
6. Non-Cell-Autonomous RSR in the CNS
7. Potential Contributions of Replicative Stress to Genomic Variations in the Nervous System
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Kermi, C.; Aze, A.; Maiorano, D. Preserving genome integrity during the early embryonic DNA replication cycles. Genes 2019, 10, 398. [Google Scholar] [CrossRef] [Green Version]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication Stress and Cancer; Nature Publishing Group: London, UK, 2015; Volume 15, pp. 276–280. [Google Scholar]
- McKinnon, P.J. Maintaining genome stability in the nervous system. Nat. Neurosci. 2013, 16, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Abugable, A.A.; Morris, J.L.M.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair 2019, 81, 102669. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, P.J. Genome integrity and disease prevention in the nervous system. Genes Dev. 2017, 31, 1180–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Katyal, S.; Downing, S.M.; Zhao, J.; Russell, H.R.; McKinnon, P.J. Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat. Neurosci. 2012, 15, 819–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.Y.; Nanjangud, G.J.; Sokolsky-Papkov, M.; Shaw, C.; Hwang, D.; Parker, J.S.; Kabanov, A.V.; Gershon, T.R. ATR maintains chromosomal integrity during postnatal cerebellar neurogenesis and is required for medulloblastoma formation. Development 2016, 143, 4038–4052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Shull, E.R.; Frappart, P.O.; Katyal, S.; Enriquez-Rios, V.; Zhao, J.; Russell, H.R.; Brown, E.J.; McKinnon, P.J. ATR maintains select progenitors during nervous system development. Embo J. 2012, 31, 1177–1189. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Schwer, B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. Dna Repair 2018, 71, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Wei, P.C.; Schwer, B. Recurrently Breaking Genes in Neural Progenitors: Potential Roles of DNA Breaks in Neuronal Function, Degeneration and Cancer. In Genome Editing in Neurosciences; Jaenisch, R., Zhang, F., Gage, F., Eds.; Springer: Berlin, Germany, 2017; pp. 63–72. [Google Scholar] [CrossRef]
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication stress, DNA Damage, inflammatory cytokines and innate immune response. Genes 2020, 11, 409. [Google Scholar] [CrossRef] [Green Version]
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA damage and innate immunity: Links and trade-offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef]
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J. Neurophysiol. 2019, 121, 1087–1091. [Google Scholar] [CrossRef]
- Barzilai, A.; Schumacher, B.; Shiloh, Y. Genome instability: Linking ageing and brain degeneration. Mech. Ageing Dev. 2017, 161, 4–18. [Google Scholar] [CrossRef]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. Eukaryotic chromosome DNA replication: Where, when, and how? Annu. Rev. Biochem. 2010, 79, 89–130. [Google Scholar] [CrossRef] [PubMed]
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA polymerases divide the labor of genome replication. Trends Cell Biol. 2016, 26, 640–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Tsegay, P.S.; Lai, Y.; Liu, Y. Replication Stress and Consequential Instability of the Genome and Epigenome. Molecules 2019, 24, 3870. [Google Scholar] [CrossRef] [Green Version]
- Mazouzi, A.; Velimezi, G.; Loizou, J.I. DNA replication stress: Causes, resolution and disease. Exp. Cell Res. 2014, 329, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Carr, A.M. Impediments to replication fork movement: Stabilisation, reactivation and genome instability. Chromosoma 2013, 122, 33–45. [Google Scholar] [CrossRef]
- Pai, C.-C.; Kearsey, S.E. A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes 2017, 8, 57. [Google Scholar] [CrossRef]
- Delfarah, A.; Parrish, S.; Junge, J.A.; Yang, J.; Seo, F.; Li, S.; Mac, J.; Wang, P.; Fraser, S.E.; Graham, N.A. Inhibition of nucleotide synthesis promotes replicative senescence of human mammary epithelial cells. J. Biol. Chem. 2019, 294, 10564–10578. [Google Scholar] [CrossRef]
- Forey, R.; Poveda, A.; Sharma, S.; Barthe, A.; Padioleau, I.; Renard, C.; Lambert, R.; Skrzypczak, M.; Ginalski, K.; Lengronne, A.; et al. Mec1 is activated at the onset of normal s phase by low-dNTP pools impeding dna replication. Mol. Cell 2020, 78, 396–410.e394. [Google Scholar] [CrossRef]
- Mathews, C.K. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 2015, 15, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, R.; Louis, T.; Robbe, P.; Ackroyd, S.; Burns, A.; Timbs, A.T.; Wright Colopy, G.; Dreau, H.; Sigaux, F.; Judde, J.G.; et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 2014, 123, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, C.; Miazzi, C.; Franzolin, E.; Pontarin, G.; Ferraro, P.; Frangini, M.; Reichard, P.; Bianchi, V. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat Res. 2010, 703, 2–10. [Google Scholar] [CrossRef]
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef] [Green Version]
- Drolet, M.; Bi, X.; Liu, L.F. Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J. Biol. Chem. 1994, 269, 2068–2074. [Google Scholar]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [Green Version]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef]
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 2016, 167, 1001–1013.e1007. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Cohen, S.; Legube, G. Transcription-Coupled DNA Double-Strand Break Repair: Active Genes Need Special Care. J. Mol. Biol. 2017, 429, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Boque-Sastre, R.; Soler, M.; Oliveira-Mateos, C.; Portela, A.; Moutinho, C.; Sayols, S.; Villanueva, A.; Esteller, M.; Guil, S. Head-to-head antisense transcription and R-loop formation promotes transcriptional activation. Proc. Natl. Acad. Sci. USA 2015, 112, 5785–5790. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, S.C.; Wang, S.; Ma, W.K.; Al Husini, N.; Dhoondia, Z.; Ansari, A.; Pascuzzi, P.E.; Tran, E.J. Regulated formation of lncRNA-DNA hybrids enables faster transcriptional induction and environmental adaptation. Mol. Cell 2016, 61, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunseich, C.; Wang, I.X.; Watts, J.A.; Burdick, J.T.; Guber, R.D.; Zhu, Z.; Bruzel, A.; Lanman, T.; Chen, K.; Schindler, A.B.; et al. senataxin mutation reveals how r-loops promote transcription by blocking DNA methylation at gene promoters. Mol. Cell 2018, 69, 426–437.e427. [Google Scholar] [CrossRef] [Green Version]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The fanconi anemia pathway protects genome integrity from r-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzminov, A. When DNA topology turns deadly - RNA polymerases dig in their r-loops to stand their ground: New positive and negative (super)twists in the replication-transcription conflict. Trends Genet. Tig. 2018, 34, 111–120. [Google Scholar] [CrossRef]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Balbo Pogliano, C.; Matos, J.; et al. Fork cleavage-religation cycle and active transcription mediate replication restart after fork stalling at co-transcriptional R-loops. Mol. Cell 2020, 77, 528–541.e528. [Google Scholar] [CrossRef]
- Wellinger, R.E.; Prado, F.; Aguilera, A. Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol. Cell Biol. 2006, 26, 3327–3334. [Google Scholar] [CrossRef] [Green Version]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; de Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates R-Loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786.e719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Alberts, B.M. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science 1995, 267, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Olavarrieta, L.; Hernández, P.; Krimer, D.B.; Schvartzman, J.B. DNA knotting caused by head-on collision of transcription and replication. J. Mol. Biol. 2002, 322, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Sankar, T.S.; Wastuwidyaningtyas, B.D.; Dong, Y.; Lewis, S.A.; Wang, J.D. The nature of mutations induced by replication–transcription collisions. Nature 2016, 535, 178–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of s phase transcription. Cell 2009, 138, 870–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [Green Version]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buj, R.; Aird, K.M. Deoxyribonucleotide triphosphate metabolism in cancer and metabolic disease. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.K. DNA precursor metabolism and genomic stability. Faseb J. 2006, 20, 1300–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discov. 2018. [Google Scholar] [CrossRef] [Green Version]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2020, 43. [Google Scholar] [CrossRef]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication Catastrophe: When a checkpoint fails because of exhaustion. Mol. Cell 2017, 66, 735–749. [Google Scholar] [CrossRef] [Green Version]
- Bastos de Oliveira, F.M.; Kim, D.; Cussiol, J.R.; Das, J.; Jeong, J.R.; Doerfler, L.; Schmidt, K.H.; Yu, H.; Smolka, M.B. Phosphoproteomics Reveals Distinct Modes of Mec1/ATR Signaling during DNA Replication. Mol. Cell 2015, 57, 1124–1132. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, T.N.; Yin, Y.; Calderon, M.J.; Qian, C.; Schamus-Haynes, S.; Sugitani, N.; Osmanbeyoglu, H.U.; Rothenberg, E.; Watkins, S.C.; Bakkenist, C.J. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc. Natl. Acad. Sci. USA 2019, 116, 13374–13383. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Ré, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 functions in break-induced replication repair of collapsed dna replication forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakofsky, C.J.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA double-strand break repair: An ever-growing toolbox. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marians, K.J. Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 2018, 87, 217–238. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Template switching: From replication fork repair to genome rearrangements. Cell 2007, 131, 1228–1230. [Google Scholar] [CrossRef] [Green Version]
- Zafar, M.K.; Eoff, R.L. Translesion DNA Synthesis in cancer: Molecular mechanisms and therapeutic opportunities. Chem. Res. Toxicol. 2017, 30, 1942–1955. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Song, X.; Ma, F.; Herrup, K. Accumulation of cytoplasmic DNA Due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J. Neurosci. 2019, 39, 6378–6394. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.W.; Herrup, K. Individual cytokines modulate the neurological symptoms of ATM Deficiency in a Region Specific Manner. eNeuro 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Baddour, J.A.; Sousounis, K.; Tsonis, P.A. Organ repair and regeneration: An overview. Birth Defects Res. C Embryo Today 2012, 96, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.L.; Dyer, M.A. Regulation of proliferation during central nervous system development. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2005. [Google Scholar] [CrossRef]
- Roussel, M.F.; Hatten, M.E. Chapter 8—Cerebellum: Development and Medulloblastoma. In Current Topics in Developmental Biology; Dyer, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 94, pp. 235–282. [Google Scholar]
- Adnani, L.; Han, S.; Li, S.; Mattar, P.; Schuurmans, C. Mechanisms of Cortical Differentiation, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 336, pp. 223–320. [Google Scholar]
- Taverna, E.; Götz, M.; Huttner, W.B. The cell biology of neurogenesis: Toward an understanding of the development and evolution of the neocortex. Advance 2014, 30, 465–502. [Google Scholar] [CrossRef] [PubMed]
- Noctor, S.C.; Flint, A.C.; Weissman, T.A.; Wong, W.S.; Clinton, B.K.; Kriegstein, A.R. Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J. Neurosci. 2002, 22, 3161–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegstein, A.; Alvarez-Buylla, A. The Glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [Green Version]
- Deneen, B.; Ho, R.; Lukaszewicz, A.; Hochstim, C.J.; Gronostajski, R.M.; Anderson, D.J. The transcription factor nfia controls the onset of gliogenesis in the developing spinal cord. Neuron 2006, 52, 953–968. [Google Scholar] [CrossRef] [Green Version]
- Bayer, S.A.; Altman, J. Neocortical Development 199, 1st ed.; Shirley, A.B., Altman, J., Eds.; Raven Press: New York, NY, USA, 1991; p. 255. [Google Scholar] [CrossRef] [Green Version]
- Kozareva, D.A.; Cryan, J.F.; Nolan, Y.M. Born this way: Hippocampal neurogenesis across the lifespan. Aging Cell 2019, 18, e13007. [Google Scholar] [CrossRef] [Green Version]
- Snyder, J.S.; Drew, M.R. Functional neurogenesis over the years. Behav. Brain Res. 2020, 382, 112470. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Allen, N.J. Astrocytes, neurons, synapses: A tripartite view on cortical circuit development. Neural Dev. 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.T.; Błasiak, J.; Niittykoski, M.; Kinnunen, K.; Kauppinen, A.; Salminen, A.; Kaarniranta, K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2017, 36, 64–77. [Google Scholar] [CrossRef]
- Blasiak, J.; Piechota, M.; Pawlowska, E.; Szatkowska, M.; Sikora, E.; Kaarniranta, K. Cellular senescence in age-related macular degeneration: Can autophagy and DNA Damage response play a role? Oxid Med. Cell Longev. 2017, 2017, 5293258. [Google Scholar] [CrossRef]
- Bassett, E.A.; Wallace, V.A. Cell fate determination in the vertebrate retina. Trends Neurosci. 2012, 35, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Cepko, C. Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci. 2014, 15, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.A.P.; Pearson, R.A. Control of cell proliferation by neurotransmitters in the developing vertebrate retina. Brain Res. 2008, 1192, 37–60. [Google Scholar] [CrossRef] [PubMed]
- Butts, T.; Green, M.J.; Wingate, R.J.T. Development of the cerebellum: Simple steps to make a ‘little brain’. Co. Biol. 2014, 141, 031–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amini, R.; Rocha-Martins, M.; Norden, C. Neuronal migration and lamination in the vertebrate retina. Front. Neurosci 2017, 11, 742. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Ming, G.L.; Song, H. Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Götz, M.; Nakafuku, M.; Petrik, D. Neurogenesis in the developing and adult brain—similarities and key differences. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Akers, K.G.; Martinez-Canabal, A.; Restivo, L.; Yiu, A.P.; De Cristofaro, A.; Hsiang, H.L.; Wheeler, A.L.; Guskjolen, A.; Niibori, Y.; Shoji, H.; et al. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science 2014, 344, 598–602. [Google Scholar] [CrossRef]
- Kempermann, G.; Gage, F.H.; Aigner, L.; Song, H.; Curtis, M.A.; Thuret, S.; Kuhn, H.G.; Jessberger, S.; Frankland, P.W.; Cameron, H.A.; et al. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 2018, 23, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Anacker, C.; Hen, R. Adult hippocampal neurogenesis and cognitive flexibility—Linking memory and mood. Nat. Rev. Neurosci 2017, 18, 335–346. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.; Rizzolo, D. Understanding medulloblastoma. J. Am. Acad. Physician Assist. 2017, 30, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Bremner, R. The search for the retinoblastoma cell of origin. Nat. Rev. Cancer 2005, 5, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [Green Version]
- Bartek Jr, J.; Fornara, O.; Merchut-Maya, J.M.; Maya-Mendoza, A.; Rahbar, A.; Stragliotto, G.; Broholm, H.; Svensson, M.; Sehested, A.; Söderberg Naucler, C.; et al. Replication stress, DNA damage signalling, and cytomegalovirus infection in human medulloblastomas. Mol. Oncol. 2017, 11, 945–964. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.P.; Gilmour, L.; et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.A.; Canman, C.E. Replication stress: An Achilles’ heel of glioma cancer stem–like cells. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbelstein, M.; Sørensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; O’Connor, M.J. Targeting the Replication Stress Response in Cancer; Elsevier: Amsterdam, The Netherlands, 2018; Volume 188, pp. 155–167. [Google Scholar]
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment; American Association for Cancer Research: Philadelphia, PA, USA, 2019; Volume 79, pp. 1730–1739. [Google Scholar]
- Yang, W.; Gao, Y. Translesion and repair DNA Polymerases: Diverse structure and mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Sugo, N.; Aratani, Y.; Nagashima, Y.; Kubota, Y.; Koyama, H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase β. Embo J. 2000, 19, 1397–1404. [Google Scholar] [CrossRef] [Green Version]
- Onishi, K.; Uyeda, A.; Shida, M.; Hirayama, T.; Yagi, T.; Yamamoto, N.; Sugo, N. Genome stability by DNA polymerase β in neural progenitors contributes to neuronal differentiation in cortical development. J. Neurosci. 2017, 37, 8444–8458. [Google Scholar] [CrossRef] [Green Version]
- Tonzi, P.; Huang, T.T. Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair 2019, 78, 20–26. [Google Scholar] [CrossRef]
- Zhuo, M.; Gorgun, M.F.; Englander, E.W. Translesion synthesis DNA polymerase kappa is indispensable for DNA repair synthesis in cisplatin exposed dorsal root ganglion neurons. Mol. Neurobiol. 2018, 55, 2506–2515. [Google Scholar] [CrossRef]
- Bochman, M.L. Roles of DNA helicases in the maintenance of genome integrity. Mol. Cell Oncol. 2014, 1, e963429. [Google Scholar] [CrossRef] [Green Version]
- Shamanna, R.A.; Croteau, D.L.; Lee, J.H.; Bohr, V.A. Recent advances in understanding werner syndrome. F1000Res 2017, 6, 1779. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Sinha, D.; Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Asaithamby, A. Werner Syndrome protein and DNA replication. Int. J. Mol. Sci. 2018, 19, 3442. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.E.; Haas, L.F. Neurological complications of Werner’s syndrome. J. Neurol. 2003, 250, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Ishikawa, Y.; Sugimoto, M.; Furuichi, Y. Werner syndrome: A changing pattern of clinical manifestations in Japan (1917~2008). Biosci. Trends 2013, 7, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, F.; Mukherjee, S.; Yang, Y.; Mori, E.; Bhattacharya, S.; Kobayashi, J.; Yannone, S.M.; Chen, D.J.; Asaithamby, A. Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 2014, 9, 1387–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannascoli, C.; Palermo, V.; Murfuni, I.; Franchitto, A.; Pichierri, P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 2015, 43, 9788–9803. [Google Scholar] [CrossRef] [Green Version]
- Palermo, V.; Rinalducci, S.; Sanchez, M.; Grillini, F.; Sommers, J.A.; Brosh, R.M.; Zolla, L.; Franchitto, A.; Pichierri, P. CDK1 phosphorylates WRN at collapsed replication forks. Nat. Commun. 2016, 7, 12880. [Google Scholar] [CrossRef]
- Kehrli, K.; Phelps, M.; Lazarchuk, P.; Chen, E.; Monnat, R., Jr.; Sidorova, J.M. Class I Histone Deacetylase HDAC1 and WRN RECQ Helicase contribute additively to protect replication forks upon Hydroxyurea-induced Arrest. J. Biol. Chem. 2016, 291, 24487–24503. [Google Scholar] [CrossRef] [Green Version]
- Cogger, V.C.; Svistounov, D.; Warren, A.; Zykova, S.; Melvin, R.G.; Solon-Biet, S.M.; O’Reilly, J.N.; McMahon, A.C.; Ballard, J.W.O.; De Cabo, R.; et al. Liver aging and pseudocapillarization in a Werner syndrome mouse model. J. Gerontol. Ser. A 2013, 69, 1076–1086. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Maierhofer, A.; Flunkert, J.; Oshima, J.; Martin, G.M.; Haaf, T.; Horvath, S. Accelerated epigenetic aging in Werner syndrome. Aging 2017, 9, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, N.; Nakamura, K.; Izumiyama-Shimomura, N.; Aida, J.; Ishii, A.; Goto, M.; Ishikawa, Y.; Asaka, R.; Matsuura, M.; Hatamochi, A.; et al. Accelerated in vivo epidermal telomere loss in Werner syndrome. Aging 2011, 3, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.W.; St-Pierre, M.K.; Detuncq, J.; Aumailley, L.; Dubois, M.J.; Couture, V.; Skuk, D.; Marette, A.; Tremblay, J.P.; Lebel, M.; et al. Nonfunctional mutant Wrn protein leads to neurological deficits, neuronal stress, microglial alteration, and immune imbalance in a mouse model of Werner syndrome. Brain Behav. Immun. 2018, 73, 450–469. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndr. 2017, 8, 4–23. [Google Scholar] [CrossRef] [PubMed]
- de Renty, C.; Ellis, N.A. Bloom’s syndrome: Why not premature aging? A comparison of the BLM and WRN helicases. Ageing Res. Rev. 2017, 33, 36–51. [Google Scholar] [CrossRef] [Green Version]
- Böhm, S.; Bernstein, K.A. The role of post-translational modifications in fine-tuning BLM helicase function during DNA repair. DNA Repair 2014, 22, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Smith, K.; Waldman, B.C.; Waldman, A.S. Depletion of the bloom syndrome helicase stimulates homology-dependent repair at double-strand breaks in human chromosomes. DNA Repair 2011, 10, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Bachrati, C.Z.; Borts, R.H.; Hickson, I.D. Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 2006, 34, 2269–2279. [Google Scholar] [CrossRef]
- Liu, Y.; Nielsen, C.F.; Yao, Q.; Hickson, I.D. The origins and processing of ultra fine anaphase DNA bridges. Curr. Opin. Genet. Dev. 2014, 26, 1–5. [Google Scholar] [CrossRef]
- Chatterjee, S.; Zagelbaum, J.; Savitsky, P.; Sturzenegger, A.; Huttner, D.; Janscak, P.; Hickson, I.D.; Gileadi, O.; Rothenberg, E. Mechanistic insight into the interaction of BLM helicase with intra-strand G-quadruplex structures. Nat. Commun. 2014, 5, 5556. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.L.; North, P.S.; Dart, A.; Lakin, N.D.; Hickson, I.D. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol. Cell Biol. 2004, 24, 1279–1291. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.A.; Conti, C.; Guirouilh-Barbat, J.; Nakamura, A.; Miao, Z.H.; Davies, S.L.; Saccá, B.; Hickson, I.D.; Bensimon, A.; Pommier, Y. Endogenous γ-H2AX-ATM-Chk2 checkpoint activation in Bloom’s syndrome helicase deficient cells is related to DNA replication arrested forks. Mol. Cancer Res. 2007, 5, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Chester, N.; Kuo, F.; Kozak, C.; O’Hara, C.D.; Leder, P. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 1998, 12, 3382–3393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larizza, L.; Roversi, G.; Volpi, L. Rothmund-Thomson syndrome. Orphanet J. Rare Dis. 2010, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinmayee, J.T.; Meghana, G.R.; Prathiba, R.K.; Ramesh, T.K. Ophthalmic manifestations in Rothmund-Thomson syndrome: Case report and review of literature. Indian J. Ophthalmol. 2017, 65, 1025–1027. [Google Scholar] [CrossRef]
- Gelaw, B.; Ali, S.; Becker, J. Rothmund-Thomson syndrome, Klippel-Feil syndrome, and osteosarcoma. Skelet. Radiol. 2004, 33, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Hameed, M.; Mandelker, D. Tumor Syndromes Predisposing to Osteosarcoma. Adv. Anat. Pathol. 2018, 25, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Shamanna, R.A.; Singh, D.K.; Lu, H.; Mirey, G.; Keijzers, G.; Salles, B.; Croteau, D.L.; Bohr, V.A. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 2014, 35, 2415–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Shamanna, R.A.; Keijzers, G.; Anand, R.; Rasmussen, L.J.; Cejka, P.; Croteau, D.L.; Bohr, V.A. RECQL4 promotes DNA end resection in repair of DNA double-strand breaks. Cell Rep. 2016, 16, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Rossi, M.L.; Singh, D.K.; Dunn, C.; Ramamoorthy, M.; Croteau, D.L.; Liu, Y.; Bohr, V.A. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J. Biol Chem. 2012, 287, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, K.; Kumano, M.; Kubota, Y.; Hashimoto, Y.; Takisawa, H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase α in the initiation of DNA replication. Mol. Cell Biol. 2006, 26, 4843–4852. [Google Scholar] [CrossRef] [Green Version]
- Sangrithi, M.N.; Bernal, J.A.; Madine, M.; Philpott, A.; Lee, J.; Dunphy, W.G.; Venkitaraman, A.R. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 2005, 121, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Shin, G.; Jeong, D.; Kim, H.; Im, J.-S.; Lee, J.-K. RecQL4 tethering on the pre-replicative complex induces unscheduled origin activation and replication stress in human cells. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Moreno-Andres, D.; Astrinidis, S.A.; Hao, Y.; Weberruss, M.; Schellhaus, A.K.; Lue, H.; Haramoto, Y.; Gruss, O.J.; Antonin, W. Chromosome alignment maintenance requires the MAP RECQL4, mutated in the Rothmund-Thomson syndrome. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Noda, T.; Furuichi, Y. Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund-Thomson syndrome caused by the mutation of DNA helicases. Nihon Yakurigaku Zasshi 2002, 119, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoki, Y.; Araki, R.; Fujimori, A.; Ohhata, T.; Koseki, H.; Fukumura, R.; Nakamura, M.; Takahashi, H.; Noda, Y.; Kito, S.; et al. Growth retardation and skin abnormalities of the Recql4-deficient mouse. Hum. Mol. Genet. 2003, 12, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.B.; Hodges, C.A.; Barnes, E.; Vogel, H.; Hassold, T.J.; Luo, G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund–Thomson syndrome. Hum. Mol. Genet. 2005, 14, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef]
- Moreira, M.-C.; Klur, S.; Watanabe, M.; Németh, A.H.; Ber, I.L.; Moniz, J.-C.; Tranchant, C.; Aubourg, P.; Tazir, M.; Schöls, L.; et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004, 36, 225–227. [Google Scholar] [CrossRef]
- Fogel, B.L.; Cho, E.; Wahnich, A.; Gao, F.; Becherel, O.J.; Wang, X.; Fike, F.; Chen, L.; Criscuolo, C.; De Michele, G.; et al. Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum. Mol. Genet. 2014, 23, 4758–4769. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.L.; La Spada, A.R.; Senataxin, A. Novel Helicase at the Interface of RNA Transcriptome Regulation and Neurobiology: From normal function to pathological roles in motor neuron disease and cerebellar degeneration. Adv. Neurobiol. 2018, 20, 265–281. [Google Scholar] [CrossRef]
- Ishiguro, T.; Taketa, K.; Gatti, R.A. Tissue of origin of elevated α-fetoprotein in ataxia-telangiectasia. Dis. Markers 1986, 4, 293–297. [Google Scholar] [PubMed]
- Stray-Pedersen, A.; Borresen-Dale, A.L.; Paus, E.; Lindman, C.R.; Burgers, T.; Abrahamsen, T.G. α fetoprotein is increasing with age in ataxia-telangiectasia. Eur. J. Paediatr. Neurol. 2007, 11, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Anheim, M.; Monga, B.; Fleury, M.; Charles, P.; Barbot, C.; Salih, M.; Delaunoy, J.P.; Fritsch, M.; Arning, L.; Synofzik, M.; et al. Ataxia with oculomotor apraxia type 2: Clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain 2009, 132, 2688–2698. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I.; Bouslam, N.; Rivaud-Péchoux, S.; Guimarães, J.; Benomar, A.; Chamayou, C.; Goizet, C.; Moreira, M.C.; Klur, S.; Yahyaoui, M.; et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: A clinical and genetic study in 18 patients. Brain 2004, 127, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Fogel, B.L.; Perlman, S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007, 6, 245–257. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Groh, M.; Albulescu, L.O.; Cristini, A.; Gromak, N. Senataxin: Genome guardian at the interface of transcription and neurodegeneration. J. Mol. Biol. 2017, 429, 3181–3195. [Google Scholar] [CrossRef]
- Bennett, C.L.; Dastidar, S.G.; Ling, S.C.; Malik, B.; Ashe, T.; Wadhwa, M.; Miller, D.B.; Lee, C.; Mitchell, M.B.; van Es, M.A.; et al. Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. 2018, 136, 425–443. [Google Scholar] [CrossRef]
- Suraweera, A.; Lim, Y.; Woods, R.; Birrell, G.W.; Nasim, T.; Becherel, O.J.; Lavin, M.F. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum. Mol. Genet. 2009, 18, 3384–3396. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Wagschal, A.; Rousset, E.; Basavarajaiah, P.; Contreras, X.; Harwig, A.; Laurent-Chabalier, S.; Nakamura, M.; Chen, X.; Zhang, K.; Meziane, O.; et al. Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell 2012, 150, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suraweera, A.; Becherel, O.J.; Chen, P.; Rundle, N.; Woods, R.; Nakamura, J.; Gatei, M.; Criscuolo, C.; Filla, A.; Chessa, L.; et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J. Cell Biol. 2007, 177, 969–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, M.F.; Gueven, N.; Grattan-Smith, P. Defective responses to DNA single- and double-strand breaks in spinocerebellar ataxia. DNA Repair 2008, 7, 1061–1076. [Google Scholar] [CrossRef] [PubMed]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin Associates with Replication Forks to Protect Fork Integrity across RNA-Polymerase-II-Transcribed Genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yüce, Ö.; West, S.C. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell Biol. 2013, 33, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Puget, N.; Lin, Y.-L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef]
- Becherel, O.J.; Yeo, A.J.; Stellati, A.; Heng, E.Y.H.; Luff, J.; Suraweera, A.M.; Woods, R.; Fleming, J.; Carrie, D.; McKinney, K.; et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet. 2013, 9, e1003435. [Google Scholar] [CrossRef] [Green Version]
- Yeo, A.J.; Becherel, O.J.; Luff, J.E.; Graham, M.E.; Richard, D.; Lavin, M.F. Senataxin controls meiotic silencing through ATR activation and chromatin remodeling. Cell Discov. 2015, 1, 15025. [Google Scholar] [CrossRef] [Green Version]
- Crow, Y.J.; Shetty, J.; Livingston, J.H. Treatments in Aicardi-Goutières syndrome. Dev. Med. Child. Neurol. 2020, 62, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; et al. Clinical and Molecular Phenotype of Aicardi-Goutières Syndrome. Am. J. Hum. Genet. 2007, 81, 713–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J.; Manel, N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Adang, L.; Gavazzi, F.; De Simone, M.; Fazzi, E.; Galli, J.; Koh, J.; Kramer-Golinkoff, J.; De Giorgis, V.; Orcesi, S.; Peer, K.; et al. Developmental outcomes of Aicardi Goutières Syndrome. J. Child. Neurol. 2020, 35, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.A.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. Part. A 2015, 167, 296–312. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Yang, Y.G.; Lindahl, T.; Barnes, D.E. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Dutta, T.; Wang, L.; Song, L.; Gu, L.; Qian, L.; Benitez, A.; Ning, S.; Malhotra, A.; Deutscher, M.P.; et al. Human DNA Exonuclease TREX1 Is Also an Exoribonuclease that acts on single-stranded RNA. J. Biol. Chem. 2015, 290, 13344–13353. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Stamp, G.; Robins, P.; Dulic, A.; Rosewell, I.; Hrivnak, G.; Daly, G.; Lindahl, T.; Barnes, D.E. Gene-targeted mice lacking the Trex1 (DNase III) 3′-->5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell Biol. 2004, 24, 6719–6727. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Du, J.; Goodier, J.L.; Hou, J.; Kang, J.; Kazazian, H.H., Jr.; Zhao, K.; Yu, X.-F. Aicardi-Goutières syndrome protein TREX1 suppresses L1 and maintains genome integrity through exonuclease-independent ORF1p depletion. Nucleic Acids Res. 2017, 45, 4619–4631. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.A.; Tejwani, L.; Trujillo, C.A.; Negraes, P.D.; Herai, R.H.; Mesci, P.; Macia, A.; Crow, Y.J.; Muotri, A.R. Modeling of TREX1-Dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell 2017, 21, 319–331.e318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Lopes, S.; Celhar, T.; Sans-Fons, G.; Serra, M.; Fairhurst, A.-M.; Lloberas, J.; Celada, A. The Exonuclease Trex1 Restrains Macrophage Proinflammatory Activation. J. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschke, K.; Achleitner, M.; Frenzel, K.; Gerbaulet, A.; Ada, S.R.; Zeller, N.; Lienenklaus, S.; Lesche, M.; Poulet, C.; Naumann, R.; et al. Loss of Trex1 in dendritic cells is sufficient to trigger systemic autoimmunity. J. Immunol. 2016, 197, 2157–2166. [Google Scholar] [CrossRef] [PubMed]
- Coggins, S.A.; Mahboubi, B.; Schinazi, R.F.; Kim, B. SAMHD1 Functions and Human Diseases. Viruses 2020, 12, 382. [Google Scholar] [CrossRef] [Green Version]
- Beloglazova, N.; Flick, R.; Tchigvintsev, A.; Brown, G.; Popovic, A.; Nocek, B.; Yakunin, A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutières syndrome and HIV-1 restriction. J. Biol. Chem. 2013, 288, 8101–8110. [Google Scholar] [CrossRef] [Green Version]
- Daddacha, W.; Koyen, A.E.; Bastien, A.J.; Head, P.E.; Dhere, V.R.; Nabeta, G.N.; Connolly, E.C.; Werner, E.; Madden, M.Z.; Daly, M.B.; et al. SAMHD1 promotes DNA end resection to facilitate DNA repair by homologous recombination. Cell Rep. 2017, 20, 1921–1935. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, A.; Martin-Fernandez, M.; Buta, S.; Kim, B.; Bogunovic, D.; Diaz-Griffero, F. SAMHD1 deficient human monocytes autonomously trigger type I interferon. Mol. Immunol. 2018, 101, 450–460. [Google Scholar] [CrossRef]
- Kretschmer, S.; Wolf, C.; König, N.; Staroske, W.; Guck, J.; Häusler, M.; Luksch, H.; Nguyen, L.A.; Kim, B.; Alexopoulou, D.; et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis 2015, 74, e17. [Google Scholar] [CrossRef] [Green Version]
- Behrendt, R.; Schumann, T.; Gerbaulet, A.; Nguyen, L.A.; Schubert, N.; Alexopoulou, D.; Berka, U.; Lienenklaus, S.; Peschke, K.; Gibbert, K.; et al. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep. 2013, 4, 689–696. [Google Scholar] [CrossRef]
- Kojima, K.; Baba, M.; Tsukiashi, M.; Nishimura, T.; Yasukawa, K. RNA/DNA structures recognized by RNase H2. Brief. Funct. Genom. 2018, 18, 169–173. [Google Scholar] [CrossRef]
- Benitez-Guijarro, M.; Lopez-Ruiz, C.; Tarnauskaitė, Ž.; Murina, O.; Mian Mohammad, M.; Williams, T.C.; Fluteau, A.; Sanchez, L.; Vilar-Astasio, R.; Garcia-Canadas, M. RNase H2, mutated in Aicardi-Goutières syndrome, promotes LINE-1 retrotransposition. Embo J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Hiller, B.; Achleitner, M.; Glage, S.; Naumann, R.; Behrendt, R.; Roers, A. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med. 2012, 209, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrendt, R.; Roers, A. Mouse models for Aicardi–Goutières syndrome provide clues to the molecular pathogenesis of systemic autoimmunity. Clin. Exp. Immunol. 2014, 175, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Lettice, L.; Tarnauskaitė, Ž.; Reddy, K.; Dix, F.; Revuelta, A.; Abbondati, E.; Rigby, R.E.; Rabe, B. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. Embo J. 2016, 35, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Pokatayev, V.; Hasin, N.; Chon, H.; Cerritelli, S.M.; Sakhuja, K.; Ward, J.M.; Morris, H.D.; Yan, N.; Crouch, R.J. RNase H2 catalytic core Aicardi-Goutières syndrome–related mutant invokes cGAS–STING innate immune-sensing pathway in mice. J. Exp. Med. 2016, 213, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, K.; Damme, M.; Regen, T.; Becker, L.; Garrett, L.; Hölter, S.M.; Knittler, K.; Borowski, C.; Waisman, A.; Glatzel, M. RNase H2 loss in murine astrocytes results in cellular defects reminiscent of nucleic acid-mediated autoinflammation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Nishikura, K. Functions and Regulation of RNA Editing by ADAR Deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [Green Version]
- Liddicoat, B.J.; Piskol., R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [Green Version]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Heale, B.S.E.; Keegan, L.P.; McGurk, L.; Michlewski, G.; Brindle, J.; Stanton, C.M.; Caceres, J.F.; O’Connell, M.A. Editing independent effects of ADARs on the miRNA/siRNA pathways. Embo J. 2009, 28, 3145–3156. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.A.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Müller, A.M.; Higuchi, M.; Seeburg, P.H. Liver Disintegration in the Mouse Embryo Caused by Deficiency in the RNA-editing Enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellåker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.; Calis, J.J.A.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Dao Thi, V.L.; Shilvock, A.R.; Hoffmann, H.H.; Rosenberg, B.R.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering translational shutdown. Cell 2018, 172, 811–824.e814. [Google Scholar] [CrossRef] [Green Version]
- Deng, P.; Khan, A.; Jacobson, D.; Sambrani, N.; McGurk, L.; Li, X.; Jayasree, A.; Hejatko, J.; Shohat-Ophir, G.; O’Connell, M.A.; et al. Adar RNA editing-dependent and -independent effects are required for brain and innate immune functions in Drosophila. Nat. Commun. 2020, 11, 1580. [Google Scholar] [CrossRef] [Green Version]
- Labrune, P.; Lacroix, C.; Goutiéres, F.; de Laveaucoupet, J.; Chevalier, P.; Zerah, M.; Husson, B.; Landrieu, P. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts. A New Progress Disorder Due Diffuse Cerebral Microangiopathy. Neurology 1996, 46, 1297. [Google Scholar] [CrossRef]
- Linnankivi, T.; Valanne, L.; Paetau, A.; Alafuzoff, I.; Hakumäki, J.M.; Kivelä, T.; Lönnqvist, T.; Mäkitie, O.; Pääkkönen, L.; Vainionpää, L.; et al. Cerebroretinal microangiopathy with calcifications and cysts. Neurology 2006, 67, 1437–1443. [Google Scholar] [CrossRef]
- Polvi, A.; Linnankivi, T.; Kivelä, T.; Herva, R.; Keating, J.P.; Mäkitie, O.; Pareyson, D.; Vainionpää, L.; Lahtinen, J.; Hovatta, I.; et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am. J. Hum. Genet. 2012, 90, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.H.; Kasher, P.R.; Mayer, J.; Szynkiewicz, M.; Jenkinson, E.M.; Bhaskar, S.S.; Urquhart, J.E.; Daly, S.B.; Dickerson, J.E.; O’Sullivan, J.; et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat. Genet. 2012, 44, 338–342. [Google Scholar] [CrossRef]
- Miyake, Y.; Nakamura, M.; Nabetani, A.; Shimamura, S.; Tamura, M.; Yonehara, S.; Saito, M.; Ishikawa, F. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol. Cell 2009, 36, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Min, J.N.; Wang, Y.; Huang, C.; Peng, T.; Chai, W.; Chang, S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. Embo J. 2012, 31, 2309–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-Y.; Majerská, J.; Lingner, J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 2013, 27, 2099–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chai, W. Pathogenic CTC1 mutations cause global genome instabilities under replication stress. Nucleic Acids Res. 2018, 46, 3981–3992. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.T.; Smogorzewska, A. SnapShot: Fanconi Anemia and Associated Proteins. Cell 2015, 160, 354.e351. [Google Scholar] [CrossRef] [Green Version]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef]
- Stivaros, S.M.; Alston, R.; Wright, N.B.; Chandler, K.; Bonney, D.; Wynn, R.F.; Will, A.M.; Punekar, M.; Loughran, S.; Kilday, J.-P.; et al. Central nervous system abnormalities in Fanconi anaemia: Patterns and frequency on magnetic resonance imaging. Br. J. Radiol. 2015, 88, 20150088. [Google Scholar] [CrossRef] [Green Version]
- Johnson-Tesch, B.A.; Gawande, R.S.; Zhang, L.; MacMillan, M.L.; Nascene, D.R. Fanconi anemia: Correlating central nervous system malformations and genetic complementation groups. Pediatr Radiol. 2017, 47, 868–876. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–r988. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., 3rd; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.Y.-C.; Tsai, S.; Aristizabal, M.J.; Wells, J.P.; Coulombe, Y.; Busatto, F.F.; Chan, Y.A.; Kumar, A.; Dan Zhu, Y.; Wang, A.Y.-H.; et al. MRE11-RAD50-NBS1 promotes Fanconi Anemia R-loop suppression at transcription–replication conflicts. Nat. Commun. 2019, 10, 4265. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Hejna, J.; Takata, M. Regulation of R-loops and genome instability in Fanconi anemia. J. Biochem. 2019, 165, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Zadorozhny, K.; Sannino, V.; Beláň, O.; Mlčoušková, J.; Špírek, M.; Costanzo, V.; Krejčí, L. Fanconi-anemia-associated mutations destabilize RAD51 Filaments and impair replication fork protection. Cell Rep. 2017, 21, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Pichierri, P.; Franchitto, A.; Rosselli, F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. Embo J. 2004, 23, 3154–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef]
- Nalepa, G.; Enzor, R.; Sun, Z.; Marchal, C.; Park, S.J.; Yang, Y.; Tedeschi, L.; Kelich, S.; Hanenberg, H.; Clapp, D.W. Fanconi anemia signaling network regulates the spindle assembly checkpoint. J. Clin. Investig. 2013, 123, 3839–3847. [Google Scholar] [CrossRef] [Green Version]
- Eppig, J.T.; Motenko, H.; Richardson, J.E.; Richards-Smith, B.; Smith, C.L. The International mouse strain resource (IMSR): Cataloging worldwide mouse and ES cell line resources. Mamm. Genome 2015, 26, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Bakker, S.T.; de Winter, J.P.; Riele, H.t. Learning from a paradox: Recent insights into Fanconi anaemia through studying mouse models. Dis. Models Mech. 2013, 6, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Houghtaling, S.; Timmers, C.; Noll, M.; Finegold, M.J.; Jones, S.N.; Meyn, M.S.; Grompe, M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003, 17, 2021–2035. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.; D’Andrea, A.; Niedernhofer, L.J. Mouse models of Fanconi anemia. Mutat. Res. 2009, 668, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Tischkowitz, M.; Winqvist, R. Using mouse models to investigate the biological and physiological consequences of defects in the Fanconi anaemia/breast cancer DNA repair signalling pathway. J. Pathol. 2011, 224, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Dubois, E.L.; Guitton-Sert, L.; Béliveau, M.; Parmar, K.; Chagraoui, J.; Vignard, J.; Pauty, J.; Caron, M.-C.; Coulombe, Y.; Buisson, R.; et al. A Fanci knockout mouse model reveals common and distinct functions for FANCI and FANCD2. Nucleic Acids Res. 2019, 47, 7532–7547. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Offit, K.; Levran, O.; Mullaney, B.; Mah, K.; Nafa, K.; Batish, S.D.; Diotti, R.; Schneider, H.; Deffenbaugh, A.; Scholl, T.; et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi Anemia. J. Natl. Cancer Inst. 2003, 95, 1548–1551. [Google Scholar] [CrossRef] [Green Version]
- Frappart, P.-O.; Lee, Y.; Lamont, J.; McKinnon, P.J. BRCA2 is required for neurogenesis and suppression of medulloblastoma. Embo J. 2007, 26, 2732–2742. [Google Scholar] [CrossRef]
- Patil, A.A.; Sayal, P.; Depondt, M.-L.; Beveridge, R.D.; Roylance, A.; Kriplani, D.H.; Myers, K.N.; Cox, A.; Jellinek, D.; Fernando, M.; et al. FANCD2 re-expression is associated with glioma grade and chemical inhibition of the Fanconi Anaemia pathway sensitises gliomas to chemotherapeutic agents. Oncotarget 2014, 5, 6414–6424. [Google Scholar] [CrossRef] [Green Version]
- Moreira, M.C.; Barbot, C.; Tachi, N.; Kozuka, N.; Uchida, E.; Gibson, T.; Mendonça, P.; Costa, M.; Barros, J.; Yanagisawa, T.; et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet. 2001, 29, 189–193. [Google Scholar] [CrossRef]
- Takashima, H.; Boerkoel, C.F.; John, J.; Saifi, G.M.; Salih, M.A.; Armstrong, D.; Mao, Y.; Quiocho, F.A.; Roa, B.B.; Nakagawa, M.; et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002, 32, 267–272. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Saifi, G.M.; Weinfeld, M.; Johansson, F.; Helleday, T.; Lupski, J.R.; Caldecott, K.W. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434, 108–113. [Google Scholar] [CrossRef]
- Shen, J.; Gilmore, E.C.; Marshall, C.A.; Haddadin, M.; Reynolds, J.J.; Eyaid, W.; Bodell, A.; Barry, B.; Gleason, D.; Allen, K.; et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet. 2010, 42, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Tebbs, R.S.; Flannery, M.L.; Meneses, J.J.; Hartmann, A.; Tucker, J.D.; Thompson, L.H.; Cleaver, J.E.; Pedersen, R.A. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev. Biol. 1999, 208, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Katyal, S.; Li, Y.; El-Khamisy, S.F.; Russell, H.R.; Caldecott, K.W.; McKinnon, P.J. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat. Neurosci. 2009, 12, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tétreault, M.; Komulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 2017, 541, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, S.; Chen, Z.; Medhurst, A.L.; Neal, J.A.; Bao, Z.; Mortusewicz, O.; McGouran, J.; Song, X.; Shen, H.; Hamdy, F.C.; et al. DNA-PKcs and PARP1 Bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2016, 76, 1078–1088. [Google Scholar] [CrossRef] [Green Version]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The importance of Poly(ADP-Ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol. Cell 2018, 71, 319–331.e313. [Google Scholar] [CrossRef] [Green Version]
- Khetarpal, P.; Das, S.; Panigrahi, I.; Munshi, A. Primordial dwarfism: Overview of clinical and genetic aspects. Mol. Genet. Genom. 2016, 291, 1–15. [Google Scholar] [CrossRef]
- Erdöl, H.; Aynaci, M.; Elmas, R.; Arslan, Y.; Imamoglu, H.I. Retinal features in Seckel’s syndrome. J. Pediatr. Ophthalmol. Strabismus 2003, 40, 299–301. [Google Scholar] [CrossRef]
- Guirgis, M.F.; Lam, B.L.; Howard, C.W. Ocular manifestations of Seckel syndrome. Am. J. Ophthalmol. 2001, 132, 596–597. [Google Scholar] [CrossRef]
- Krzyżanowska-Berkowska, P.; Szumny, D.; Młyńczak, T.; Kisza, K.; Oficjalska, J. Bilateral retinal detachment in Seckel syndrome. Can. J. Ophthalmol. 2014, 49, e130–e131. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, D.; Bae, B.I.; Walsh, C.A. The Genetics of Primary Microcephaly. Annu. Rev. Genom. Hum. Genet. 2018, 19, 177–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia–telangiectasia and Rad3–related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Mokrani-Benhelli, H.; Gaillard, L.; Biasutto, P.; Le Guen, T.; Touzot, F.; Vasquez, N.; Komatsu, J.; Conseiller, E.; Pïcard, C.; Gluckman, E.; et al. Primary microcephaly, impaired DNA replication, and genomic instability caused by compound heterozygous ATR mutations. Hum. Mutat. 2013, 34, 374–384. [Google Scholar] [CrossRef]
- Ogi, T.; Walker, S.; Stiff, T.; Hobson, E.; Limsirichaikul, S.; Carpenter, G.; Prescott, K.; Suri, M.; Byrd, P.J.; Matsuse, M.; et al. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genet. 2012, 8, e1002945. [Google Scholar] [CrossRef] [Green Version]
- Yazinski, S.A.; Zou, L. Functions, regulation, and therapeutic implications of the ATR checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar]
- de Klein, A.; Muijtjens, M.; van Os, R.; Verhoeven, Y.; Smit, B.; Carr, A.M.; Lehmann, A.R.; Hoeijmakers, J.H.J. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000, 10, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Murga, M.; Bunting, S.; Montaña, M.F.; Soria, R.; Mulero, F.; Cañamero, M.; Lee, Y.; McKinnon, P.J.; Nussenzweig, A.; Fernandez-Capetillo, O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009, 41, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Amirifar, P.; Ranjouri, M.R.; Yazdani, R.; Abolhassani, H.; Aghamohammadi, A. Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatr. Allergy Immunol. 2019, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houldsworth, J.; Lavin, M.F. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980, 8, 3709–3720. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.B.; Young, B.R. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc. Natl. Acad. Sci. USA 1980, 77, 7315–7317. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Armata, H.L.; Golebiowski, D.; Jung, D.Y.; Ko, H.J.; Kim, J.K.; Sluss, H.K. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol. Cell Biol. 2010, 30, 5787–5794. [Google Scholar] [CrossRef] [Green Version]
- Valentin-Vega, Y.A.; Maclean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.R.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Ousset, M.; Bouquet, F.; Fallone, F.; Biard, D.; Dray, C.; Valet, P.; Salles, B.; Muller, C. Loss of ATM positively regulates the expression of hypoxia inducible factor 1 (HIF-1) through oxidative stress: Role in the physiopathology of the disease. Cell Cycle 2010, 9, 2814–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Eaton, J.S.; Lin, Z.P.; Sartorelli, A.C.; Bonawitz, N.D.; Shadel, G.S. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Investig. 2007, 117, 2723–2734. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Saintigny, Y.; Delacôte, F.; Varès, G.; Petitot, F.; Lambert, S.; Averbeck, D.; Lopez, B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. Embo J. 2001, 20, 3861–3870. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Enriquez-Rios, V.; Dumitrache, L.C.; Downing, S.M.; Li, Y.; Brown, E.J.; Russell, H.R.; McKinnon, P.J. DNA-PKcs, ATM, and ATR Interplay Maintains Genome Integrity during Neurogenesis. J. Neurosci. 2017, 37, 893–905. [Google Scholar] [CrossRef]
- van der Burg, M.; Ijspeert, H.; Verkaik, N.S.; Turul, T.; Wiegant, W.W.; Morotomi-Yano, K.; Mari, P.-O.; Tezcan, I.; Chen, D.J.; Zdzienicka, M.Z.; et al. A DNA-PKcs mutation in a radiosensitive T–B– SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Investig. 2009, 119, 91–98. [Google Scholar] [CrossRef]
- Woodbine, L.; Neal, J.A.; Sasi, N.-K.; Shimada, M.; Deem, K.; Coleman, H.; Dobyns, W.B.; Ogi, T.; Meek, K.; Davies, E.G.; et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J. Clin. Investig. 2013, 123, 2969–2980. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.J.; Chi, L.; So, S.; Lee, K.-J.; Mori, E.; Fattah, K.; Yang, J.; Chen, D.J. BRCA1 modulates the autophosphorylation status of DNA-PKcs in S phase of the cell cycle. Nucleic Acids Res. 2014, 42, 11487–11501. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.; Ye, R.; Trinkle-Mulcahy, L.; Neal, J.A.; De Wever, V.; Morrice, N.A.; Meek, K.; Lees-Miller, S.P. Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci. Rep. 2014, 34. [Google Scholar] [CrossRef] [PubMed]
- Espejel, S.; Franco, S.; Sgura, A.; Gae, D.; Bailey, S.M.; Taccioli, G.E.; Blasco, M.A. Functional interaction between DNA-PKcs and telomerase in telomere length maintenance. Embo J. 2002, 21, 6275–6287. [Google Scholar] [CrossRef] [Green Version]
- Fisher, T.S.; Zakian, V.A. Ku: A multifunctional protein involved in telomere maintenance. DNA Repair 2005, 4, 1215–1226. [Google Scholar] [CrossRef]
- Huang, B.; Shang, Z.F.; Li, B.; Wang, Y.; Liu, X.D.; Zhang, S.M.; Guan, H.; Rang, W.Q.; Hu, J.A.; Zhou, P.K. DNA-PKcs associates with PLK1 and is involved in proper chromosome segregation and cytokinesis. J. Cell Biochem. 2014, 115, 1077–1088. [Google Scholar] [CrossRef]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G2-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef]
- Serrano, M.A.; Li, Z.; Dangeti, M.; Musich, P.R.; Patrick, S.; Roginskaya, M.; Cartwright, B.; Zou, Y. DNA-PK, ATM and ATR collaboratively regulate p53–RPA interaction to facilitate homologous recombination DNA repair. Oncogene 2013, 32, 2452–2462. [Google Scholar] [CrossRef] [Green Version]
- Shang, Z.; Yu, L.; Lin, Y.F.; Matsunaga, S.; Shen, C.Y.; Chen, B.P. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis 2014, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Shao, R.G.; Cao, C.X.; Zhang, H.; Kohn, K.W.; Wold, M.S.; Pommier, Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. Embo J. 1999, 18, 1397–1406. [Google Scholar] [CrossRef]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yajima, H.; Lee, K.J.; Chen, B.P. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol. Cell Biol. 2006, 26, 7520–7528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-F.; Shih, H.-Y.; Shang, Z.; Matsunaga, S.; Chen, B.P.C. DNA-PKcs is required to maintain stability of Chk1 and Claspin for optimal replication stress response. Nucleic Acids Res. 2014, 42, 4463–4473. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Shih, H.Y.; Shang, Z.F.; Kuo, C.T.; Guo, J.; Du, C.; Lee, H.; Chen, B.P.C. PIDD mediates the association of DNA-PKcs and ATR at stalled replication forks to facilitate the ATR signaling pathway. Nucleic Acids Res. 2018, 46, 1847–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, T.; Gell, D.; Fox, M.; Taccioli, G.E.; Lehmann, A.R.; Jackson, S.P.; Jeggo, P.A. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc. Natl. Acad. Sci. USA 1996, 93, 10285–10290. [Google Scholar] [CrossRef] [Green Version]
- Bogue, M.A.; Jhappan, C.; Roth, D.B. Analysis of variable (diversity) joining recombination in DNAdependent protein kinase (DNA-PK)-deficient mice reveals DNA-PK-independent pathways for both signal and coding joint formation. Proc. Natl. Acad. Sci. USA 1998, 95, 15559–15564. [Google Scholar] [CrossRef] [Green Version]
- Goytisolo, F.A.; Samper, E.; Edmonson, S.; Taccioli, G.E.; Blasco, M.A. The absence of the dna-dependent protein kinase catalytic subunit in mice results in anaphase bridges and in increased telomeric fusions with normal telomere length and G-strand overhang. Mol. Cell Biol. 2001, 21, 3642–3651. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Sekiguchi, J.; Gao, Y.; Dikkes, P.; Frank, K.; Ferguson, D.; Hasty, P.; Chun, J.; Alt, F.W. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 2668–2673. [Google Scholar] [CrossRef] [Green Version]
- Niimi, N.; Sugo, N.; Aratani, Y.; Koyama, H. Genetic interaction between DNA polymerase β and DNA-PKcs in embryogenesis and neurogenesis. Cell Death Differ. 2005, 12, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Vemuri, M.C.; Schiller, E.; Naegele, J.R. Elevated DNA double strand breaks and apoptosis in the CNS of scid mutant mice. Cell Death Differ. 2001, 8, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Baleriola, J.; Suárez, T.; de la Rosa, E.J. DNA-PK promotes the survival of young neurons in the embryonic mouse retina. Cell Death Differ. 2010, 17, 1697–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, A.; Tainer, J.A. The MRE11–RAD50–NBS1 Complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell 2018, 71, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Zaki-Dizaji, M.; Akrami, S.M.; Azizi, G.; Abolhassani, H.; Aghamohammadi, A. Inflammation, a significant player of Ataxia–Telangiectasia pathogenesis? Inflamm. Res. 2018, 67, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Casari, E.; Rinaldi, C.; Marsella, A.; Gnugnoli, M.; Colombo, C.V.; Bonetti, D.; Longhese, M.P. Processing of DNA double-strand breaks by the MRX complex in a chromatin context. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Fiévet, A.; Bellanger, D.; Valence, S.; Mobuchon, L.; Afenjar, A.; Giuliano, F.; Dubois d’Enghien, C.; Parfait, B.; Pedespan, J.M.; Auger, N.; et al. Three new cases of ataxia-telangiectasia-like disorder: No impairment of the ATM pathway, but S-phase checkpoint defect. Hum. Mutat. 2019, 40, 1690–1699. [Google Scholar] [CrossRef]
- Taylor, A.M.R.; Groom, A.; Byrd, P.J. Ataxia-telangiectasia-like disorder (ATLD)—its clinical presentation and molecular basis. DNA Repair 2004, 3, 1219–1225. [Google Scholar] [CrossRef]
- Theunissen, J.-W.F.; Kaplan, M.I.; Hunt, P.A.; Williams, B.R.; Ferguson, D.O.; Alt, F.W.; Petrini, J.H.J. Checkpoint failure and chromosomal instability without Lymphomagenesis in Mre11ATLD1/ATLD1 Mice. Mol. Cell 2003, 12, 1511–1523. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Heil, C.; Sahún-Roncero, M.; Colicchia, V.; Veschi, V.; Albini, S.; Fruci, D.; Ricci, B.; Soriani, A.; et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. 2016, 23, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Baple, E.L.; Chambers, H.; Cross, H.E.; Fawcett, H.; Nakazawa, Y.; Chioza, B.A.; Harlalka, G.V.; Mansour, S.; Sreekantan-Nair, A.; Patton, M.A.; et al. Hypomorphic PCNA mutation underlies a human DNA repair disorder. J. Clin. Investig. 2014, 124, 3137–3146. [Google Scholar] [CrossRef] [Green Version]
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Bagińska, B.; Kalina, M.A.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Primers 2019, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Petersen, S.; Tessarollo, L.; Nussenzweig, A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 2001, 11, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.R.; Mirzoeva, O.K.; Morgan, W.F.; Lin, J.; Dunnick, W.; Petrini, J.H.J. A murine model of Nijmegen Breakage Syndrome. Curr. Biol. 2002, 12, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Baranes, K.; Raz-Prag, D.; Nitzan, A.; Galron, R.; Ashery-Padan, R.; Rotenstreich, Y.; Assaf, Y.; Shiloh, Y.; Wang, Z.Q.; Barzilai, A.; et al. Conditional inactivation of the NBS1 gene in the mouse central nervous system leads to neurodegeneration and disorganization of the visual system. Exp. Neurol. 2009, 218, 24–32. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Grigaravicius, P.; Remus, M.; Cavalheiro, G.R.; Gomes, A.L.; Rocha-Martins, M.; Frappart, L.; Reuss, D.; McKinnon, P.J.; von Deimling, A.; et al. Nbn and atm cooperate in a tissue and developmental stage-specific manner to prevent double strand breaks and apoptosis in developing brain and eye. PLoS ONE 2013, 8, e69209. [Google Scholar] [CrossRef]
- Dar, I.; Yosha, G.; Elfassy, R.; Galron, R.; Wang, Z.Q.; Shiloh, Y.; Barzilai, A. Investigation of the functional link between ATM and NBS1 in the DNA damage response in the mouse cerebellum. J. Biol. Chem. 2011, 286, 15361–15376. [Google Scholar] [CrossRef] [Green Version]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Chiu, Y.-H.; Chen, Z.J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.J.; LeBert, N.; Chitre, A.A.; Koo, C.X.E.; Nga, X.H.; Ho, S.S.W.; Khatoo, M.; Tan, N.Y.; Ishii, K.J.; Gasser, S. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. 2015, 11, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammert, C.R.; Frost, E.L.; Bellinger, C.E.; Bolte, A.C.; McKee, C.A.; Hurt, M.E.; Paysour, M.J.; Ennerfelt, H.E.; Lukens, J.R. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 2020, 580, 647–652. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hui, C.W.; Li, J.; Herrup, K. The Interaction of the Atm Genotype with Inflammation and Oxidative Stress. PLoS ONE 2014, 9, e85863. [Google Scholar] [CrossRef]
- Hui, C.W.; Song, X.; Ma, F.; Shen, X.; Herrup, K. Ibuprofen prevents progression of ataxia telangiectasia symptoms in ATM-deficient mice. J. Neuroinflammation 2018, 15, 308. [Google Scholar] [CrossRef] [Green Version]
- Quek, H.; Luff, J.; Cheung, K.; Kozlov, S.; Gatei, M.; Lee, C.S.; Bellingham, M.C.; Noakes, P.G.; Lim, Y.C.; Barnett, N.L.; et al. A rat model of ataxia-telangiectasia: Evidence for a neurodegenerative phenotype. Hum. Mol. Genet. 2016, 26, 109–123. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, Y.; Frank, K.M.; Dikkes, P.; Fujiwara, Y.; Seidl, K.J.; Sekiguchi, J.M.; Rathbun, G.A.; Swat, W.; Wang, J.; et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 1998, 95, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Frank, K.M.; Sekiguchi, J.M.; Seidl, K.J.; Swat, W.; Rathbun, G.A.; Cheng, H.L.; Davidson, L.; Kangaloo, L.; Alt, F.W. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 1998, 396, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.T.; Kaushal, D.; Murphy, M.; Zhang, Y.; Datta, A.; Chen, C.; Monroe, B.; Mostoslavsky, G.; Coakley, K.; Gao, Y.; et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7378–7383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Meyers, R.M. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 2016, 11, 853–871. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.C.; Lee, C.S.; Du, Z.; Schwer, B.; Zhang, Y.; Kao, J.; Zurita, J.; Alt, F.W. Three classes of recurrent DNA break clusters in brain progenitors identified by 3D proximity-based break joining assay. Proc. Natl. Acad. Sci. USA 2018, 115, 1919–1924. [Google Scholar] [CrossRef] [Green Version]
- Arlt, M.F.; Wilson, T.E.; Glover, T.W. Replication stress and mechanisms of CNV formation. Curr. Opin. Genet. Dev. 2012, 22, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Glover, T.W.; Wilson, T.E. Molecular biology: Breaks in the brain. Nature 2016, 532, 46–47. [Google Scholar] [CrossRef]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Weissman, I.L.; Gage, F.H. A Mechanism for Somatic Brain Mosaicism. Cell 2016, 164, 593–595. [Google Scholar] [CrossRef] [Green Version]
- D’Gama, A.M.; Walsh, C.A. Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 2018, 21, 1504–1514. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. The ageing genome, clonal mosaicism and chronic disease. Curr. Opin. Genet. Dev. 2017, 42, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, M.J.; Moran, J.V.; Abyzov, A.; Akbarian, S.; Bae, T.; Cortes-Ciriano, I.; Erwin, J.A.; Fasching, L.; Flasch, D.A.; Freed, D.; et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 2017, 356, eaal1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrback, S.; Siddoway, B.; Liu, C.S.; Chun, J. Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 2018, 78, 1026–1048. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M.A.; Woodworth, M.B.; Lee, S.; Evrony, G.D.; Mehta, B.K.; Karger, A.; Lee, S.; Chittenden, T.W.; D’Gama, A.M.; Cai, X.; et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015, 350, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, T.; Tomasini, L.; Mariani, J.; Zhou, B.; Roychowdhury, T.; Franjic, D.; Pletikos, M.; Pattni, R.; Chen, B.-J.; Venturini, E.; et al. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 2018, 359, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Karaca, E.; Harel, T.; Pehlivan, D.; Jhangiani, S.N.; Gambin, T.; Coban Akdemir, Z.; Gonzaga-Jauregui, C.; Erdin, S.; Bayram, Y.; Campbell, I.M.; et al. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Russell, T.A.; Grubisha, M.J.; Remmers, C.L.; Kang, S.K.; Forrest, M.P.; Smith, K.R.; Kopeikina, K.J.; Gao, R.; Sweet, R.A.; Penzes, P. A Schizophrenia-Linked KALRN Coding Variant Alters Neuron Morphology, Protein Function, and Transcript Stability. Biol. Psychiatry 2018, 83, 499–508. [Google Scholar] [CrossRef]
- Wiszniewski, W.; Gawlinski, P.; Gambin, T.; Bekiesinska-Figatowska, M.; Obersztyn, E.; Antczak-Marach, D.; Akdemir, Z.H.C.; Harel, T.; Karaca, E.; Jurek, M.; et al. Comprehensive genomic analysis of patients with disorders of cerebral cortical development. Eur. J. Hum. Genet. 2018, 26, 1121–1131. [Google Scholar] [CrossRef]
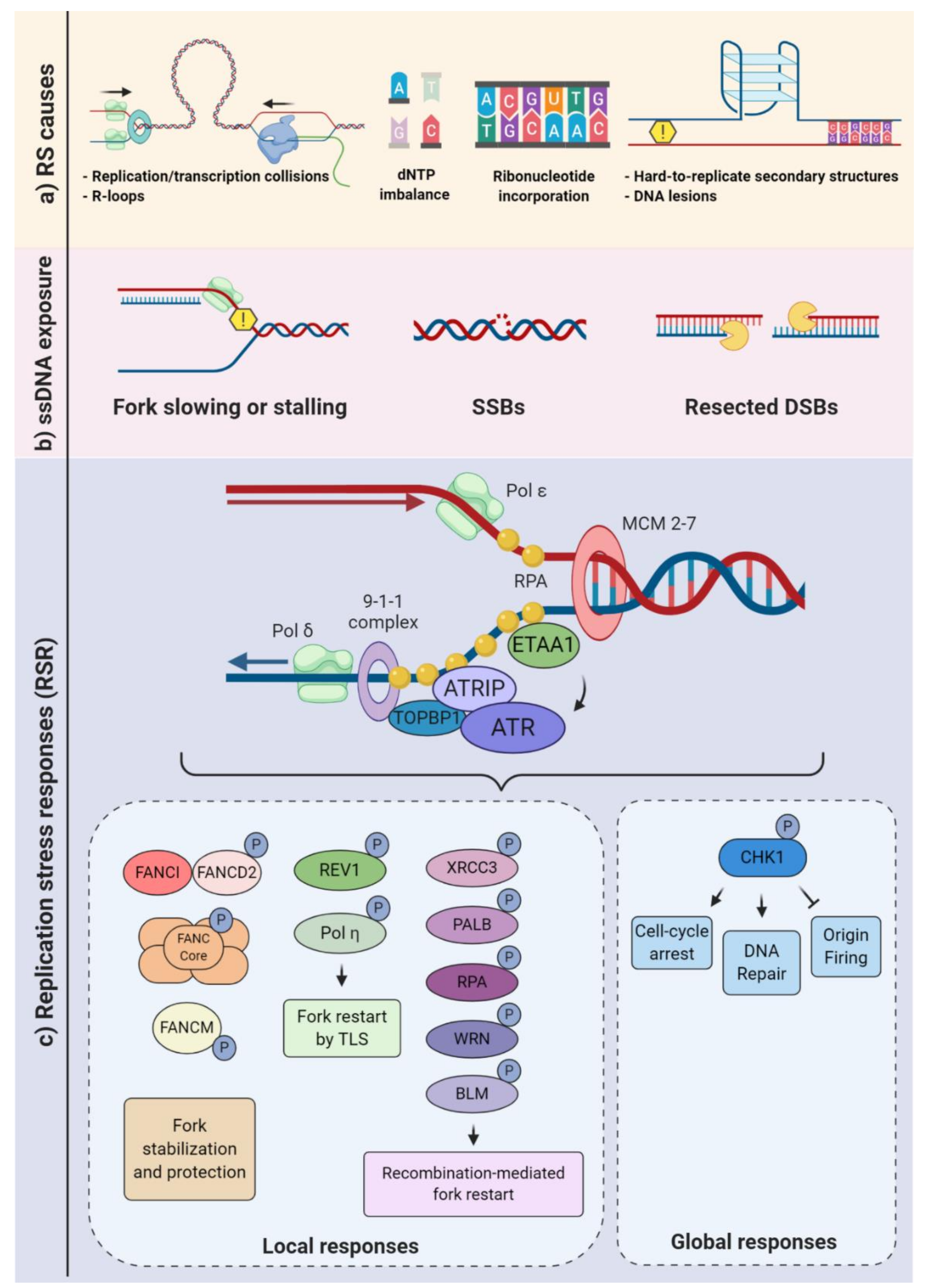
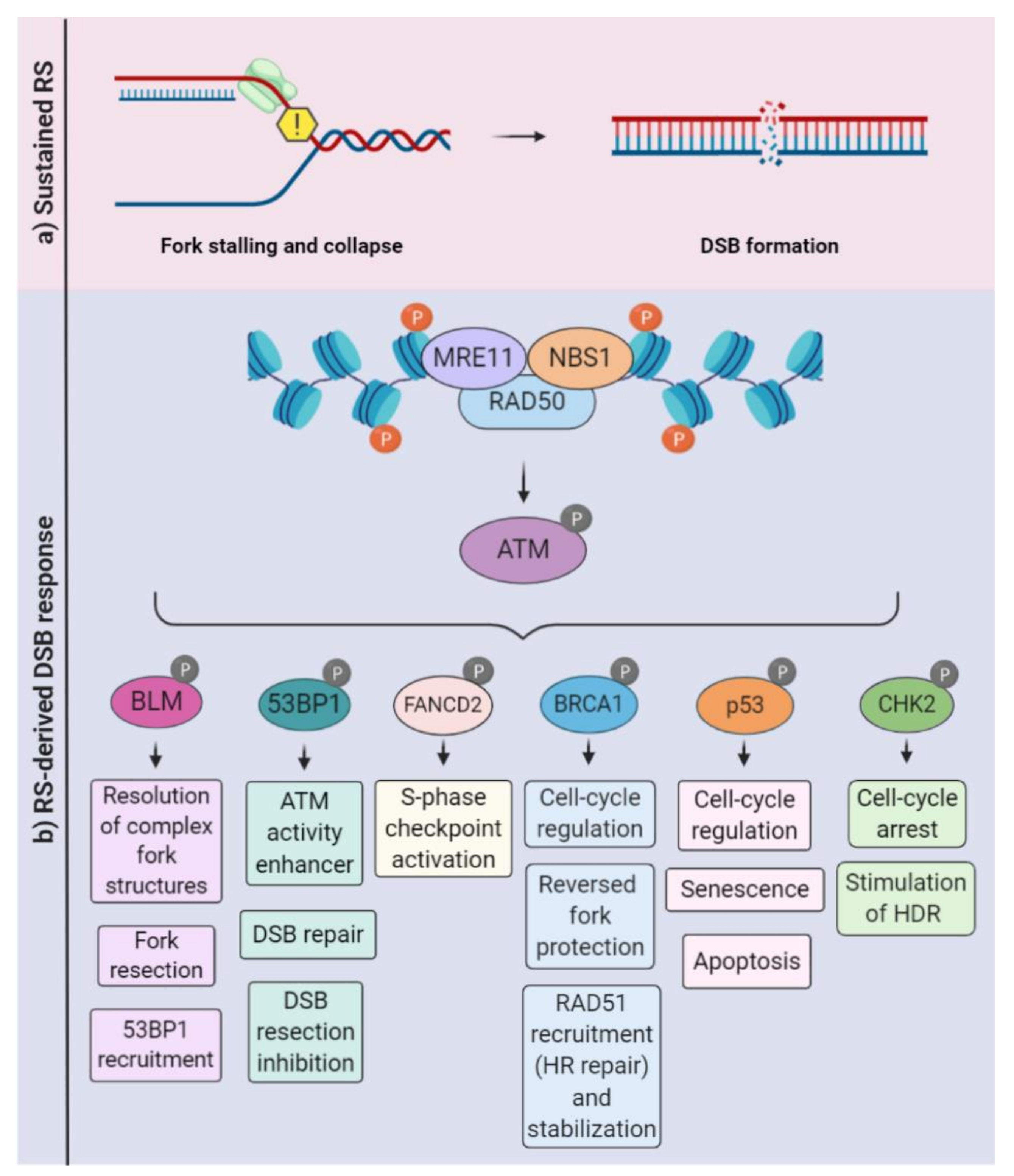


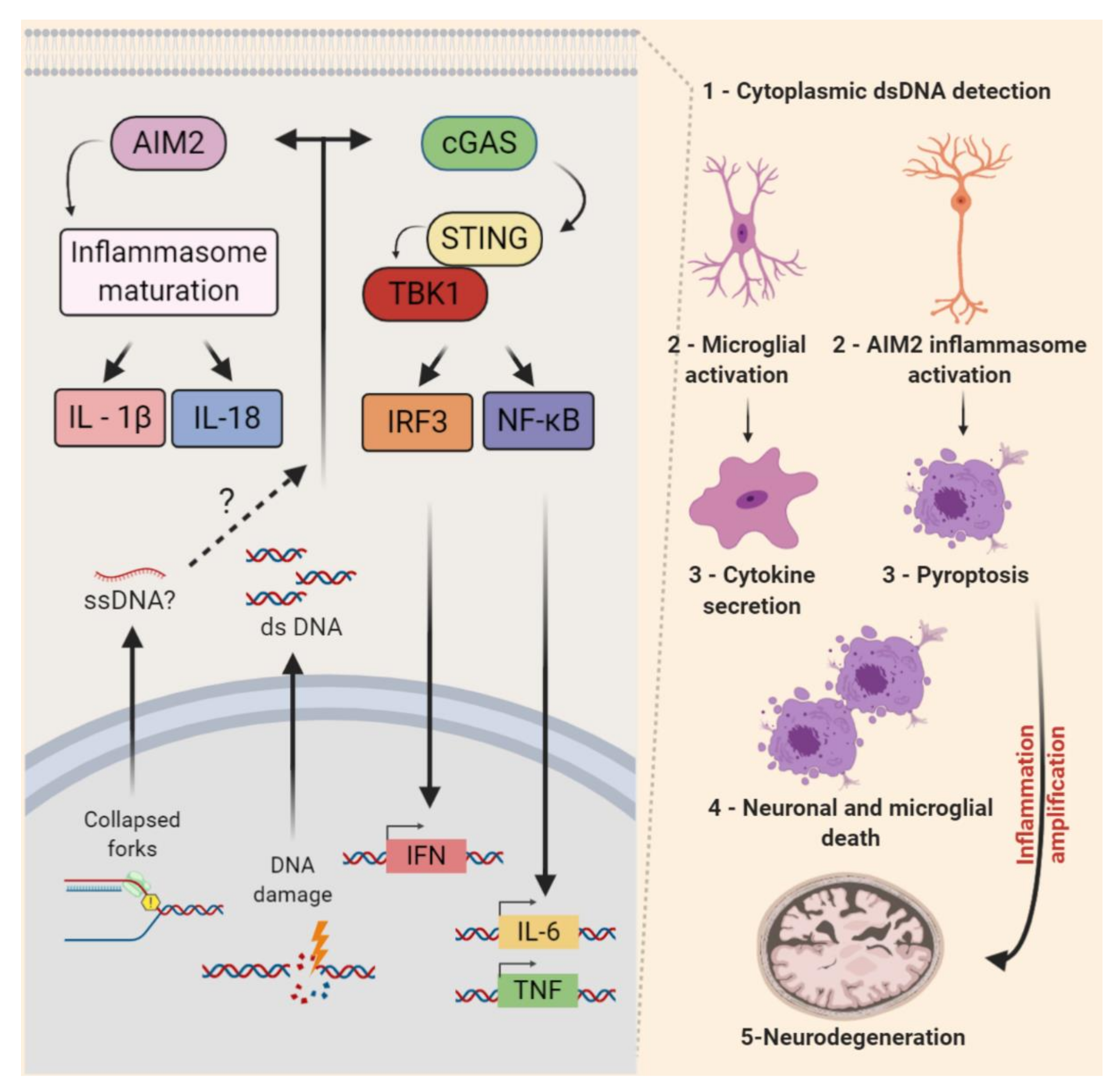

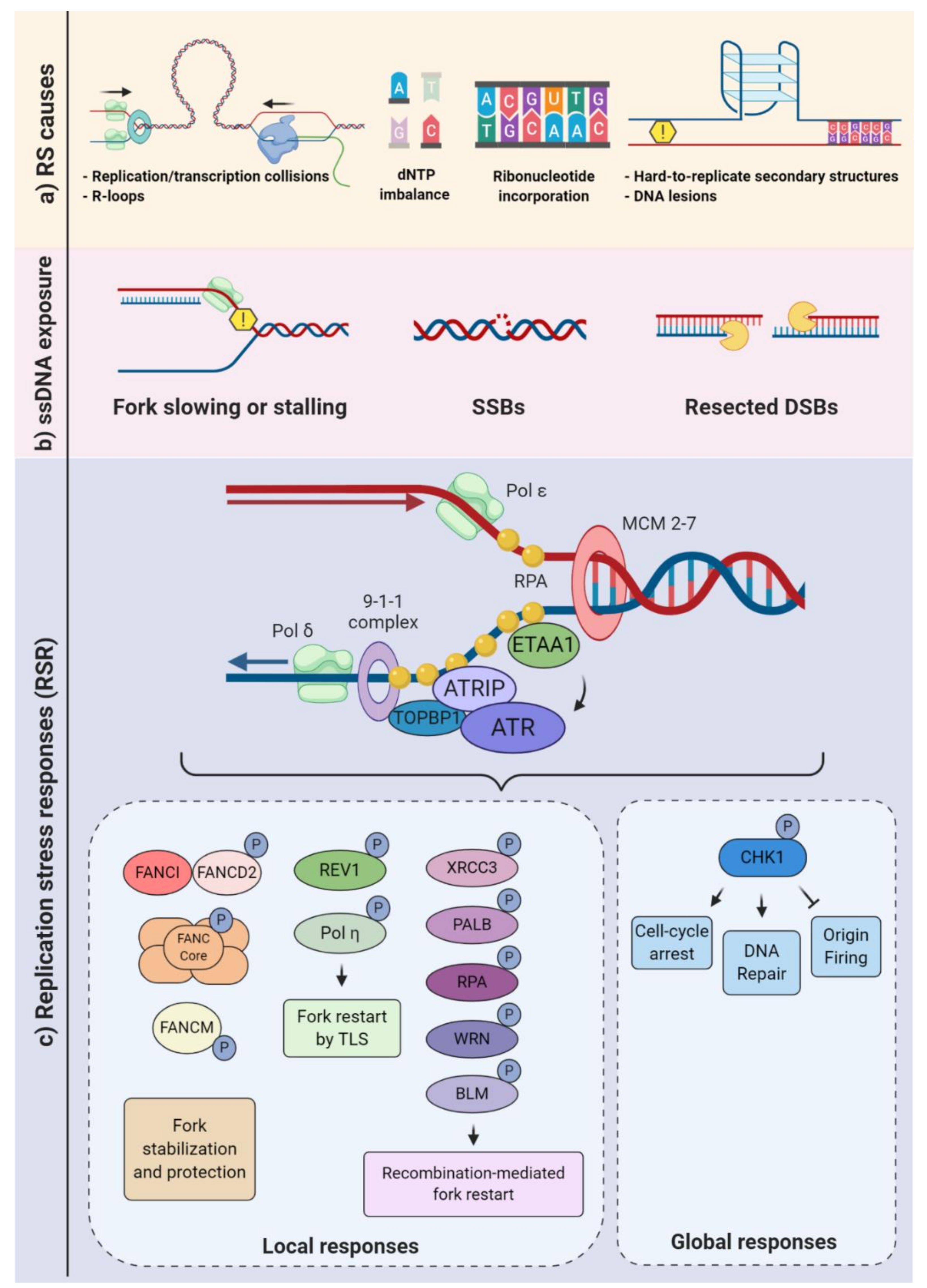{kind=link}
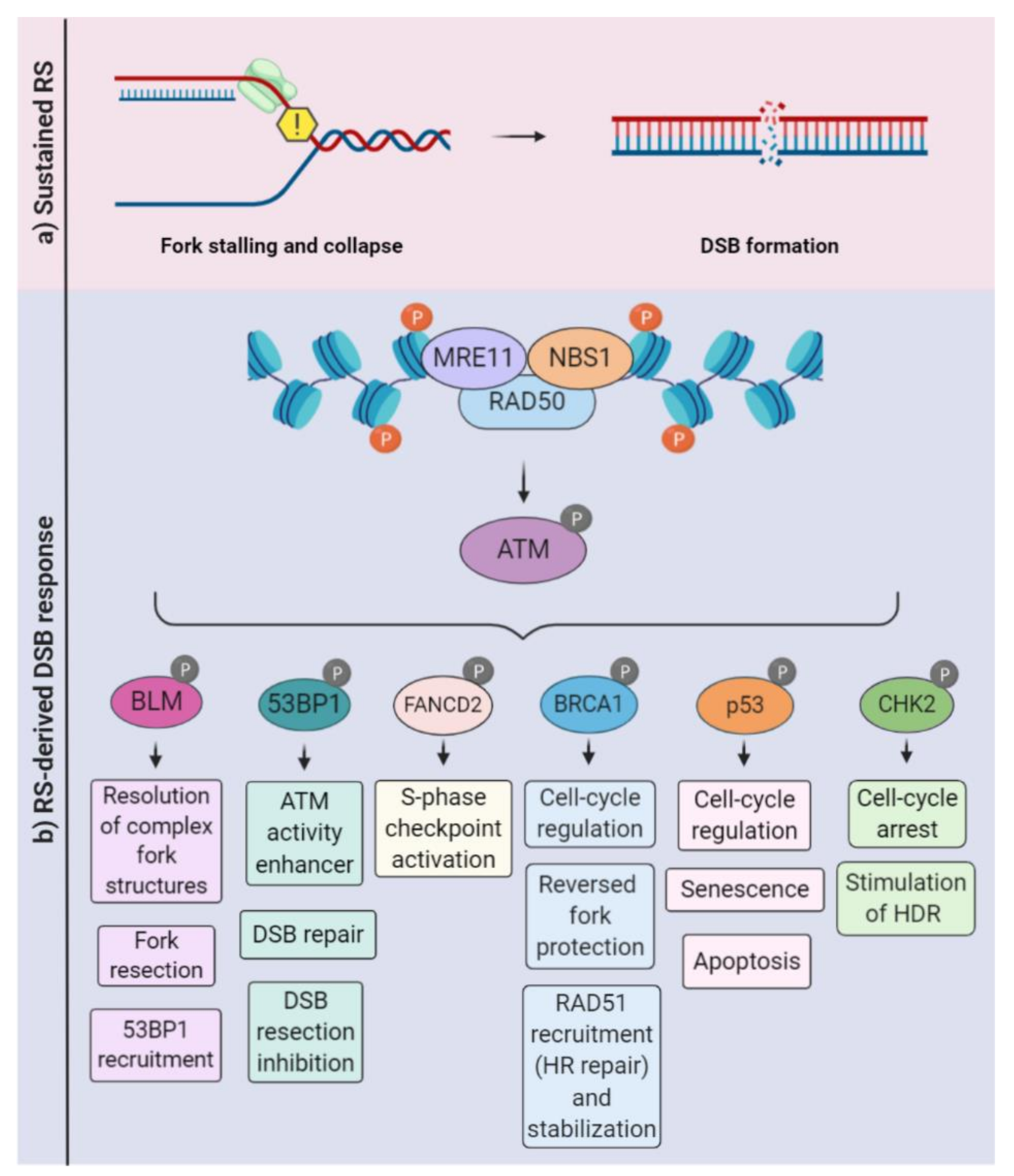{kind=link}
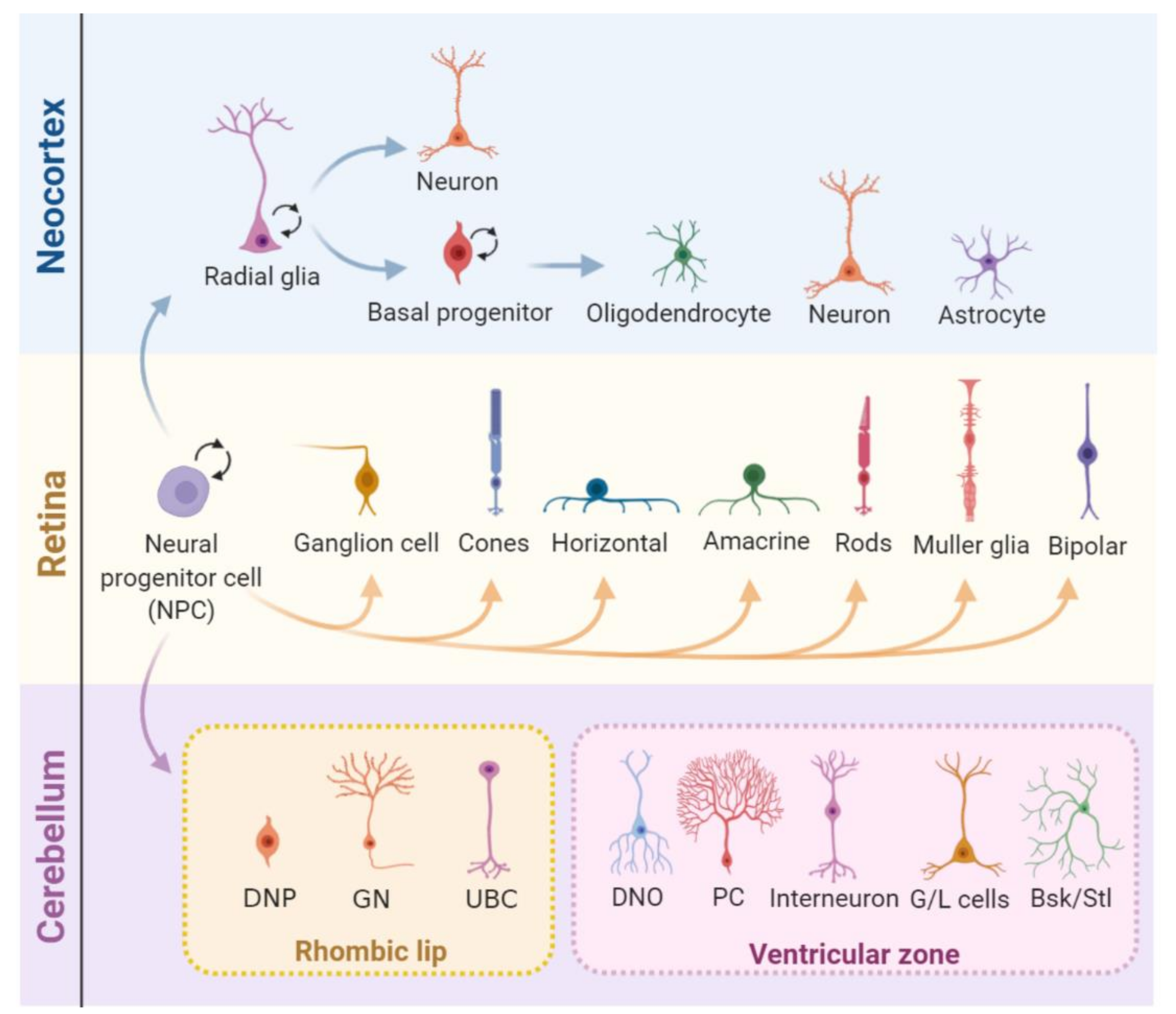{kind=link}
{kind=link}
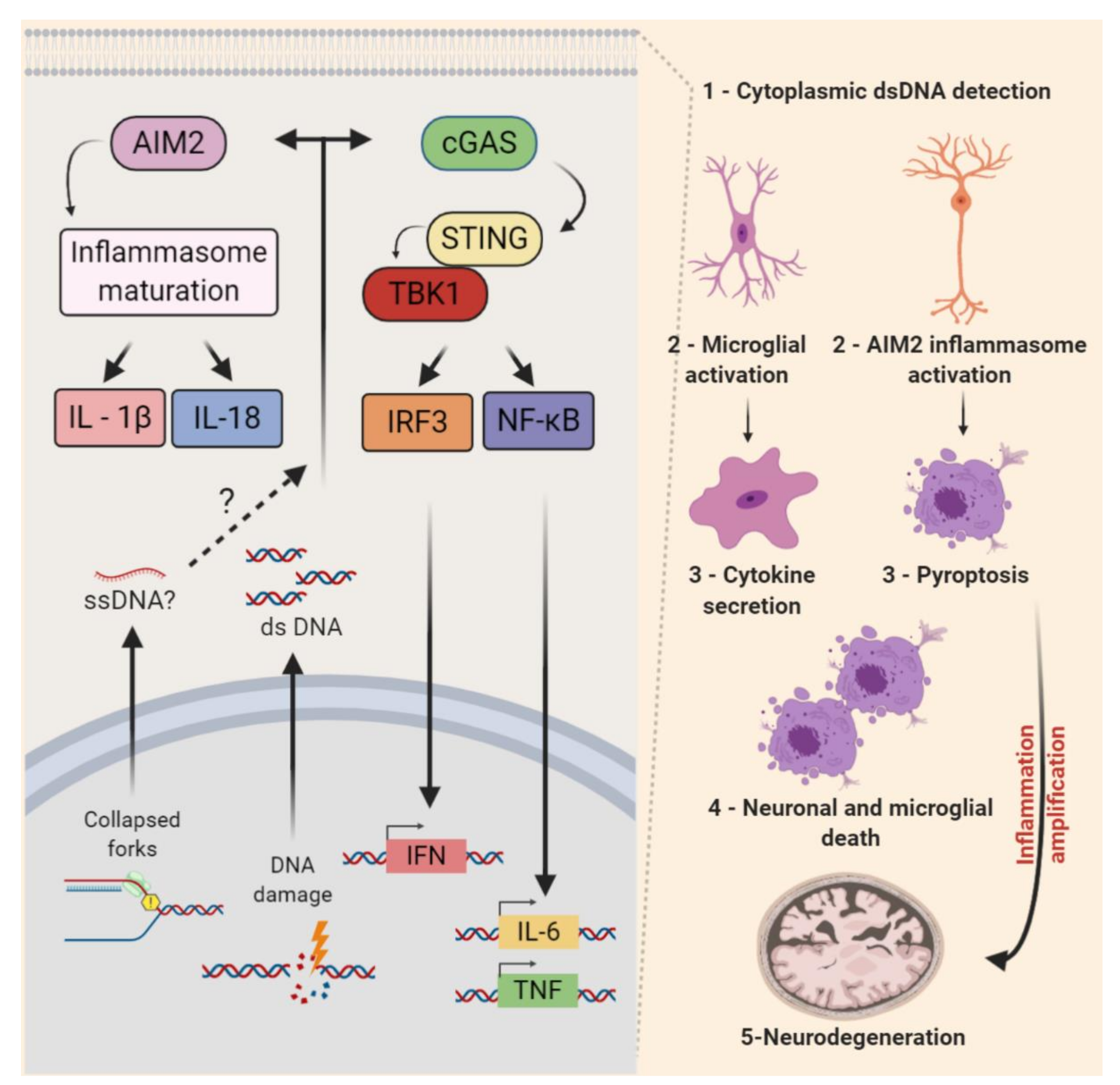{kind=link}
| Syndrome | OMIM | Mutated Genes | Mechanisms Described | Neuropathology |
|---|---|---|---|---|
| Seckel Syndrome (SS) | 210600 | ATR, ATRIP | Replication stress response | Microcephaly, cortical and retinal malformations |
| Ataxia telangiectasia (A-T) | 208900 | ATM | DSB signaling, Replication stress | Neurodegeneration |
| Ataxia-telangiectasia-like disorder 1 (ATLD1) | 604391 | MRE11 | DSB signaling, Fork resection | Ataxia, neurodegeneration, dysarthria, oculomotor apraxia |
| Nijmegen Breakage Syndrome (NBS) | 251260 | NBN | DSB signaling | Microcephaly |
| Fanconi Anemia (FA) | 605724 227646 | BRCA2, FANCD2 | DNA crosslink repair Fork protection | Medulloblastoma, microcephaly and hydrocephalus |
| Aicardi-Goutières Syndrome (AGS) | 225750 610181 610329 610333 612952 615010 | TREX1 RNASEH2B RNASEH2C RNASEH2A SAMHD1 ADAR1 | Removal of DNA:RNA hybrids, Ribonucleotide excision, dNTP hydrolysis, RNA editing | Microcephaly, cerebral atrophy, demyelination |
| Werner Syndrome (WRN) | 277700 | WRN | Complex fork structure resolution | Brain atrophy, memory deficits |
| Immunodeficiency 26 (IM26) | 615966 | PRKDC | DSB signaling | Brain atrophy, hypomyelination, visual impairment, hearing loss |
| Spinocerebellar ataxia, autosomal recessive 26 (SCAR26) | 617633 | XRCC1 | Scaffold protein | Ocular motor apraxia, progressive cerebellar atrophy |
| Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 2 | 606002 | SETX (AR) * | DNA/RNA helicase | Cerebellar ataxia, axonal neuropathy, oculomotor apraxia |
| Amyotrophic lateral sclerosis 4 | 602433 | SETX (AD) * | DNA/RNA helicase | Spinal cord atrophy, hyperreflexia, axonal neuropathy |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forrer Charlier, C.; Martins, R.A.P. Protective Mechanisms Against DNA Replication Stress in the Nervous System. Genes 2020, 11, 730. https://doi.org/10.3390/genes11070730
Forrer Charlier C, Martins RAP. Protective Mechanisms Against DNA Replication Stress in the Nervous System. Genes. 2020; 11(7):730. https://doi.org/10.3390/genes11070730
Chicago/Turabian StyleForrer Charlier, Clara, and Rodrigo A. P. Martins. 2020. "Protective Mechanisms Against DNA Replication Stress in the Nervous System" Genes 11, no. 7: 730. https://doi.org/10.3390/genes11070730
APA StyleForrer Charlier, C., & Martins, R. A. P. (2020). Protective Mechanisms Against DNA Replication Stress in the Nervous System. Genes, 11(7), 730. https://doi.org/10.3390/genes11070730