PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Clinical Characterization of the Study Subjects
2.3. Genetic Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic basis of inherited retinal disease in a molecularly characterized cohort of over 3000 families from the United Kingdom. Ophthalmology 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Chakraborty, D.; Conly, S.M.; Zulliger, R.; Naash, M.I. The K153Del PROH2 mutation differentially impacts photoreceptor structure and function. Hum. Mol. Genet. 2016, 25, 3500–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://www.retina-international.org/files/sci-news//rdsmut.htm (accessed on 28 May 2020).
- Conley, S.M.; Naash, M.I. Focus on molecules: RDS. Exp. Eye Res. 2009, 89, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.; Cideciyan, A.V.; Maguire, A.V.; Bennet, J.; Sheffield, V.C.; Stone, E.M. Preferential rod and cone photoreceptor abnormalities in heterozygotes with point mutations in the RDS gene. Exp. Eye Res. 1996, 63, 603–608. [Google Scholar] [CrossRef]
- Apfelstedt-Sylla, E.; Theischen, M.; Rüther, K.; Wedemann, H.; Gal, A.; Zrenner, E. Extensive intrafamilial and interfamilial phenotypic variation among patients with autosomal dominant retinal dystrophy and mutations in the human RDS/peripherin gene. Br. J. Ophthalmol. 1995, 79, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Boon, C.J.; Hollander, A.I.D.; Hoyng, C.B.; Cremers, F.P.; Klevering, B.J.; Keunen, J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2014, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, Unit7.20. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Coco-Martín, R.; Tellena, J.J.; Sanabria, M.R.; Rodríguez-Rúa, E.; Garcia, M.T. PRPH2 (Peripherin/RDS) Mutations Associated with Different Macular Dystrophies in a Spanish Population: A New Mutation. Eur. J. Ophthalmol. 2010, 20, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 2014, 17, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, F.; Marini, V.; Rossi, S.; Interlandi, E.; Nesti, A.; Rinaldi, M.; Varano, M.; Garrè, C.; Simonelli, F. A novel mutation in the RDS gene in an Italian family with pattern dystrophy. Br. J. Ophthalmol. 2005, 89, 1066–1068. [Google Scholar] [CrossRef] [Green Version]
- Yanagihashi, S.; Nakazawa, M.; Kurotaki, J.; Sato, M.; Miyagawa, Y.; Ohguro, H. Autosomal Dominant Central Areolar Choroidal Dystrophy and a Novel Arg195Leu Mutation in the Peripherin/RDS Gene. Arch. Ophthalmol. 2003, 121, 1458–1461. [Google Scholar] [CrossRef] [Green Version]
- Fishman, G.A.; Stone, E.M.; Gilbert, L.D.; VanDenburgh, K.; Sheffield, V.C.; Heckenlively, J.R. Clinical Features of a Previously Undescribed Codon 216 (proline to serine) Mutation in the Peripherin/Retinal Degeneration Slow Gene in Autosomal Dominant Retinitis Pigmentosa. Ophthalmology 1994, 101, 1409–1421. [Google Scholar] [CrossRef]
- Weleber, R.G.; Carr, R.E.; Murphey, W.H.; Sheffield, V.C.; Stone, E.M. Phenotypic Variation Including Retinitis Pigmentosa, Pattern Dystrophy, and Fundus Flavimaculatus in a Single Family with a Deletion of Codon 153 or 154 of the Peripherin/RDS Gene. Arch. Ophthalmol. 1993, 111, 1531–1542. [Google Scholar] [CrossRef]
- Wells, J.; Wroblewski, J.; Keen, J.; Inglehearn, C.; Jubb, C.; Eckstein, A.; Jay, M.; Arden, G.; Bhattacharya, S.; Fitzke, F.; et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat. Genet. 1993, 3, 213–218. [Google Scholar] [CrossRef]
- Travis, G.H.; Hepler, J.E. A medley of retinal dystrophies. Nat. Genet. 1993, 3, 191–192. [Google Scholar] [CrossRef]
- Boon, C.J.F.; van Schooneveld, M.J.; den Hollander, A.I.; van Lith-Verhoeven, J.J.C.; Zonneveld-Vrieling, M.N.; Theelen, T.; Cremers, F.P.M.; Hoyng, C.B.; Klevering, B.J. Mutations in the peripherin/RDS gene are an important cause of multifocal patter dystrophy simulating STGD1/fundus flavimaculatus. Br. J. Ophthalmol. 2007, 91, 1504–1511. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.F. Role of Peripherin/rds in Vertebrate Photoreceptor Architecture and Inherited Retinal Degenerations. Int. Rev. Cytol. 2006, 253, 131–175. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Lewis, T.R.; Makia, M.S.; Kakakhel, M.; Al-Ubaidi, M.R.; Arshavsky, V.Y.; Naash, M.I. Photoreceptor disc enclosure occurs in the absence of normal peripherin-2/rds oligomerization. Front. Cell. Neurosci. 2020, 28, 14–92. [Google Scholar] [CrossRef]
- Chakraborty, D.; Strayve, D.G.; Makia, M.S.; Conley, S.M.; Kakahel, M.; Al-Ubaidi, M.R.; Naash, M.I. Novel molecular mechanisms for Prph2-associated pattern dystrophy. FASEB J. 2019, 34, 1211–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheetz, T.E.; Kim, K.-Y.; Swiderski, R.E.; Philp, A.R.; Braun, T.A.; Knudtson, K.L.; Dorrance, A.M.; DiBona, G.F.; Huang, J.; Casavant, T.L.; et al. Regulation of gene expression in the mammalian eye and its relevance to eye disease. Proc. Natl. Acad. Sci. USA 2006, 103, 14429–14434. [Google Scholar] [CrossRef] [Green Version]
- Leroy, B.P.; Kailasanathan, A.; De Laey, J.-J.; Black, G.C.; Manson, F. Intrafamilial phenotypic variability in families with RDS mutations: Exclusion of ROM1 as a genetic modifier for those with retinitis pigmentosa. Br. J. Ophthalmol. 2006, 91, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samardzija, M.; Wenzel, A.; Naash, M.; Reme, C.E.; Grimm, C. Rpe65 as a modifier gene for inherited retinal degeneration. Eur. J. Neurosci. 2006, 23, 1028–1034. [Google Scholar] [CrossRef] [Green Version]
- Poloschek, C.; Bach, M.; Lagreze, W.A.; Glaus, E.; Lemke, J.R.; Berger, W.; Neidhardt, J. ABCA4andROM1: Implications for Modification of thePRPH2-Associated Macular Dystrophy Phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDs and ROM1: Historical context current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef] [Green Version]
- Tebbe, L.; Kakakhel, M.; Makia, M.S.; Al-Ubaidi, M.R.; Naash, M.I. The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases. Cells 2020, 9, 784. [Google Scholar] [CrossRef] [Green Version]
- Nichols, B.E.; Sheffield, V.C.; VanDenburgh, K.; Drack, A.V.; Kimura, A.E.; Stone, E.M. Butterfly–shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nat. Genet. 1993, 3, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Gamundi, M.J.; Hernan, I.; Muntanyola, M.; Trujillo, M.J.; García-Sandoval, B.; Ayuso, C.; Baiget, M.; Carballo, M. High prevalence of mutations in peripherin/RDS in autosomal dominant macular dystrophies in a Spanish population. Mol. Vis. 2007, 13, 1031–1037. [Google Scholar] [PubMed]
- Keilhauer, C.N.; Meigen, T.; Weber, B.H.F. Clinical Findings in a Multigeneration Family With Autosomal Dominant Central Areolar Choroidal Dystrophy Associated With an Arg195Leu Mutation in the Peripherin/RDS Gene. Arch. Ophthalmol. 2006, 124, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Paskowitz, D.M.; Lavail, M.M.; Duncan, J.L. Light and inherited retinal degeneration. Br. J. Ophthalmol. 2006, 90, 1060–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Despriet, M.D.G.; Klaver, C.C.W.; Witteman, J.C.; Bergen, A.A.; Kardys, I.; De Maat, M.P.M.; Boekhoorn, S.S.; Vingerling, J.R.; Hofman, A.; Oostra, B.A.; et al. Complement Factor H Polymorphism, Complement Activators, and Risk of Age-Related Macular Degeneration. JAMA 2006, 296, 301. [Google Scholar] [CrossRef] [Green Version]
- Berson, E.L.; Rosner, B.A.; Sandberg, M.A.; Weigel-DiFranco, C.; Moser, A.; Brockhurst, R.J.; Hayes, K.C.; Johnson, C.A.; Anderson, E.J.; Gaudio, A.R.; et al. Clinical Trial of Docosahexaenoic Acid in Patients with Retinitis PigmentosaReceiving Vitamin A Treatment. Arch. Ophthalmol. 2004, 122, 1297–1305. [Google Scholar] [CrossRef]
- Zampaglione, E.; Kinde, B.; Emily, M.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 2020, 22, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.D.; Wheaton, D.K.; Bowne, S.J.; Sullivan, L.S.; Birch, D.G.; Chen, R.; Daiger, S.P. Next-generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes contributing to disease in extended families. Mol. Vis. 2017, 23, 470–481. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient (Gene Variant) | Gender | Age at Onset | Symptoms at Onset | BCVA at First Visit (Age) | BCVA at Last Visit (Age) | Visual Field | ffERG |
|---|---|---|---|---|---|---|---|
| F1-II-2 p.Arg46Ter | M | 56 | Metamorphopsia followed by central vision loss | 20/32 RE; 20/25 LE (56) | N/A | Small central scotoma | Normal |
| F1-II-3 p.Arg46Ter | Fe | 51 | Metamorphopsia | 20/25 BE (51) | N/A | Normal | Normal |
| F1-II-4 p.Arg46Ter | M | 46 | Metamorphopsia | 20/25 BE (46) | N/A | Normal | Normal |
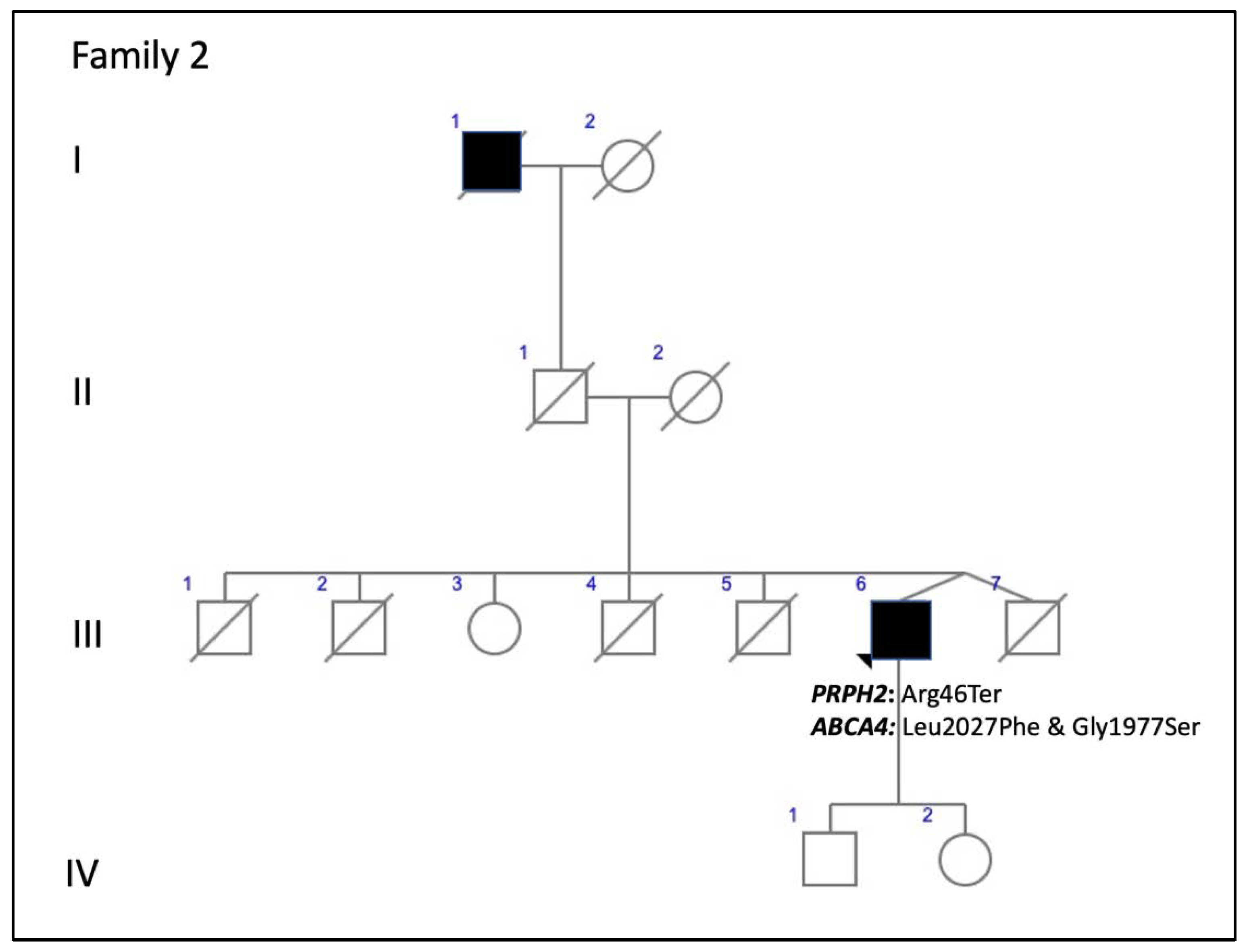
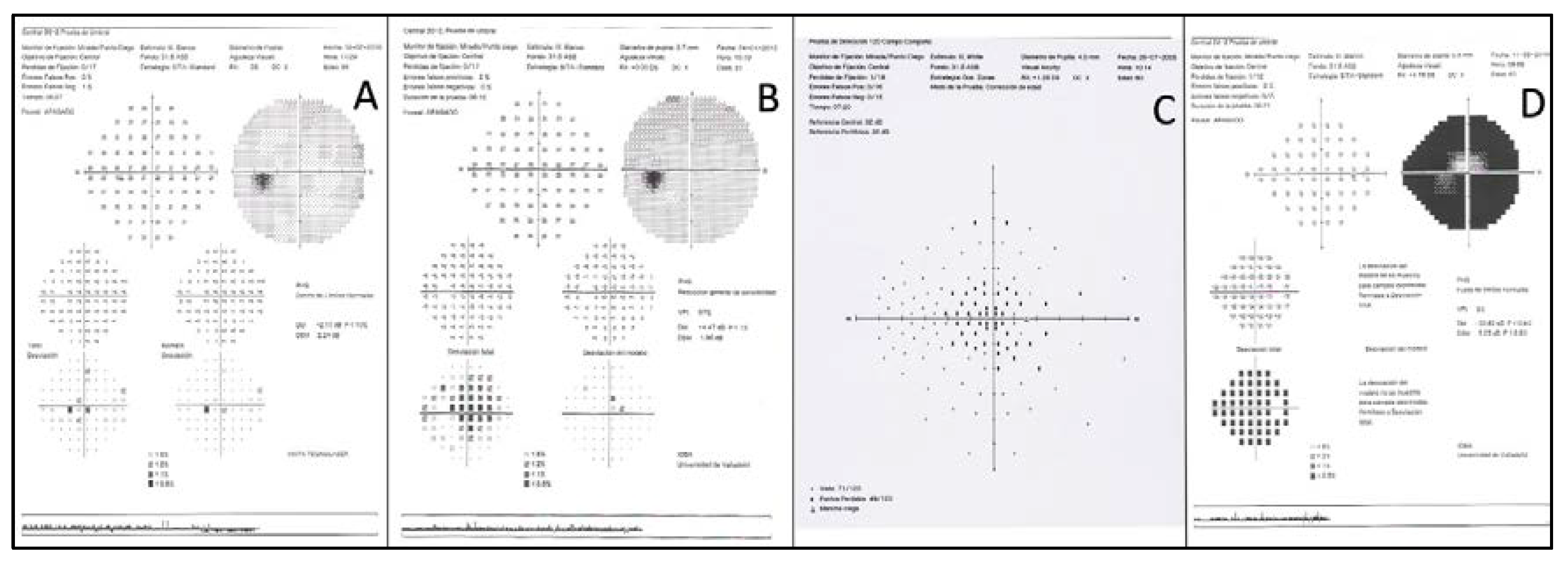
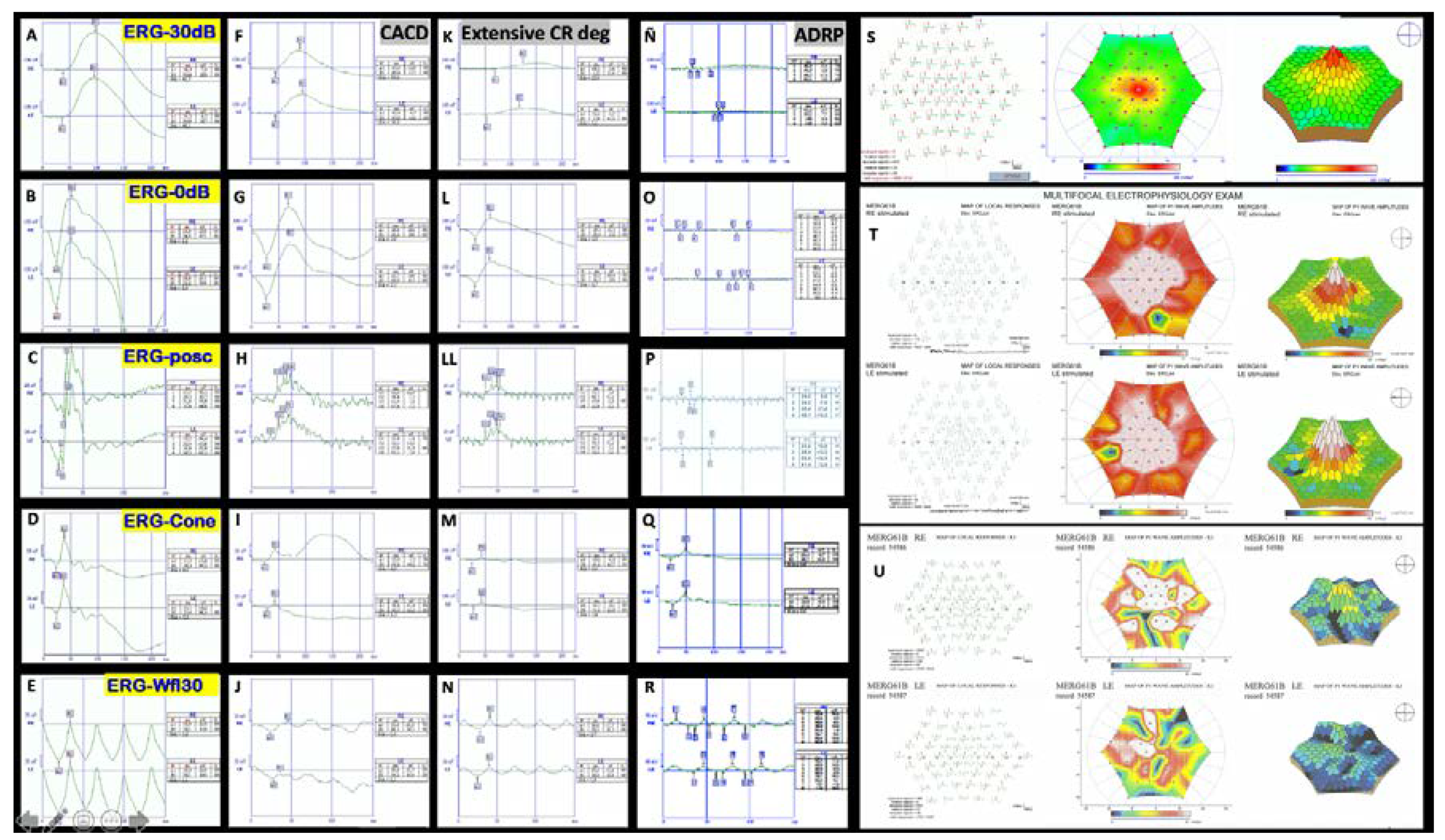
| F2-III-6 PRPH2: p.Arg46Ter ABCA4: p.Leu2027Phe/p.Gly1977Ser | M | 52 | Loss of peripheral visual field | 20/50 RE; 20/40 LE (61) | 20/63 RE/20/200 LE (76) | Large central scotoma | Scotopic and photopic subnormal 0 dB b-wave amplitude = 152 μV RE/182 μV LE; photopic = 22 μV RE/28 μV LE |
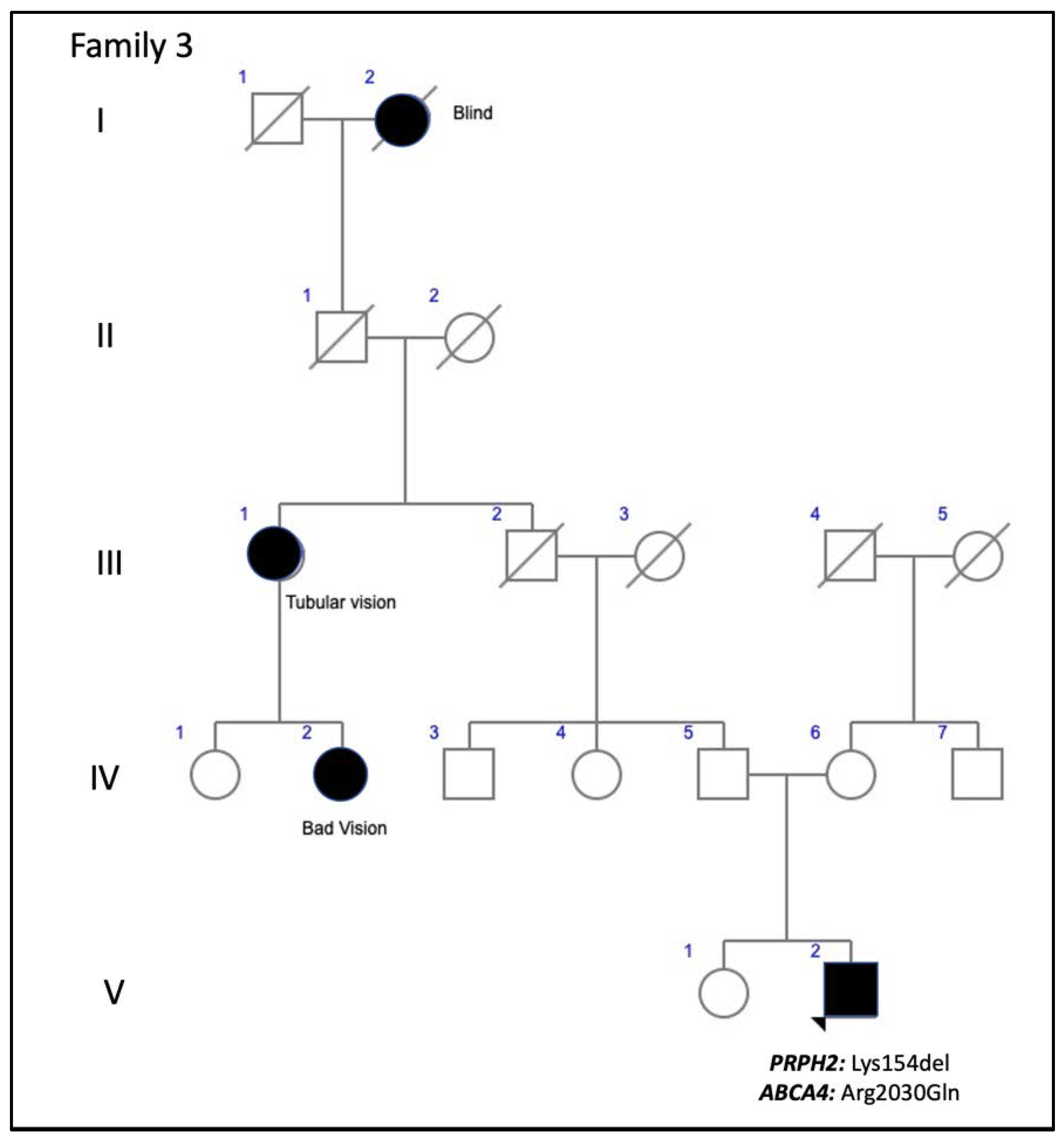
| F3-V-2 PRPH2: p.Lys154del ABCA4: p.Arg2030Gln | M | 37 | Metamorphopsia | 20/16 RE; 20/20 LE (37) | N/A | Small paracentral scotomas | Scotopic subnormal and photopic preserved 0 dB b-wave amplitude = 222 μV RE/219 μV LE |
| F4-III-1 p.Gly167Ser | Fe | 68 | Casual finding after cataract surgery | 20/25 BE (68) | 20/100 RE; 20/32 LE (78) | Large central scotoma | Scotopic abolished and photopic subnormal Photopic b-wave amplitude = 22 μV RE/28 μV LE |
| F4-IV-1 p.Gly167Ser | Fe | 53 | Asymptomatic, casual finding after Dx of her mother | 20/20 BE (53) | N/A | Paracentral scotoma | Scotopic and photopic subnormal 0 dB b-wave amplitude = 231 μV RE/218 μV LE; photopic = 57 μV RE/61 μV LE |
| F4-IV-2 p.Gly167Ser | Fe | 51 | Asymptomatic, casual finding after Dx of her mother | 20/20 BE (51) | N/A | Normal | Normal |
| F5a-III-2 p.Arg195Leu | M | 53 | Loss of central vision | 20/32 RE; 20/100 LE (59) | N/A | N/A | Normal |
| F5a-III-7 p.Arg195Leu | M | 58 | Asymptomatic, casual finding after Dx of his brother | 20/20 RE; 20/32 LE (58) | N/A | N/A | Normal |
| F5a-III-8 p.Arg195Leu | M | 59 | Asymptomatic, casual finding after Dx of his brother | 20/125 RE; 20/100 LE (59) | N/A | N/A | Normal |
| F5a-III-10 p.Arg195Leu | M | 42 | Loss of central vision | 20/63 RE; 20/25 LE (55) | 20/400 RE; 20/50 LE (61) | Central scotoma | Normal |
| F5b-III-4 p.Arg195Leu | Fe | 26 | Loss of central vision | 20/400 BE (89) | N/A | Large central scotoma | Scotopic subnormal and photopic abolished 0 dB b-wave amplitude = 87 μV RE/113 μV LE |
| F5b-IV-1 p.Arg195Leu | M | 39 | Loss of central vision | 20/25 RE; 20/32 LE (42) | N/A | Central scotoma | Scotopic and photopic subnormal 0 dB b-wave amplitude = 228 μV RE/169 μV LE; photopic = 27 μV RE/28 μV LE |
| F5b-IV-2 p.Arg195Leu | Fe | 31 | Asymptomatic, casual finding after Dx of his cousin | 20/20 BE (31) | N/A | Small central scotoma | Scotopic and photopic subnormal 0 dB b-wave amplitude = 256 μV RE/253 μV LE; photopic = 25 μV RE/27 μV LE |
| F5b-IV-4 p.Arg195Leu | M | 37 | Loss of central vision | 20/40 RE; 20/25 LE (37) | 20/63 RE/20/25 LE (50) | Large central scotoma | Scotopic subnormal and photopic abolished 0 dB b-wave amplitude = 174 μV RE/216 μV LE |
| F5c-IV-4 p.Arg195Leu | Fe | 25 | Loss of central vision | 20/800 RE/CF LE (77) | N/A | Central scotoma | Scotopic subnormal and photopic abolished 0 dB b-wave amplitude = 231 μV RE/218 μV LE |
| F5c-V-2 p.Arg195Leu | Fe | 35 | Loss of central vision | 20/200 RE; 20/125 LE (41) | N/A | Central scotoma | Scotopic subnormal and photopic abolished 0 dB b-wave amplitude = 251 μV RE/259 μV LE |
| F5c-VI-2 p.Arg195Leu | Fe | 18 | Loss of central vision | 20/50 RE; 20/32 LE (18) | N/A | central scotoma | Scotopic subnormal and photopic abolished 0 dB b-wave amplitude = 258 μV RE/299 μV LE; |
| F5d-VI-1 p.Arg195Leu | M | 20 | Loss of central vision and night blindness | 20/32 BE (25) | 20/100 BE (45) | Large central scotoma | Scotopic abolished and photopic subnormal Photopic b-wave amplitude = 35 μV RE/38 μV LE |
| F6-I-5 p.Val209Ile | M | 56 | Metamorphopsia followed by centralvision loss | 20/32 RE; 20/25 LE (62) | 20/400 BE (76) | First normal, then central scotoma | Normal |
| 6-II-2 p.Val209Ile | Fe | 51 | Asymptomatic, casual finding | 20/20 BE (50) | 20/20 BE (54) | Normal | Normal |
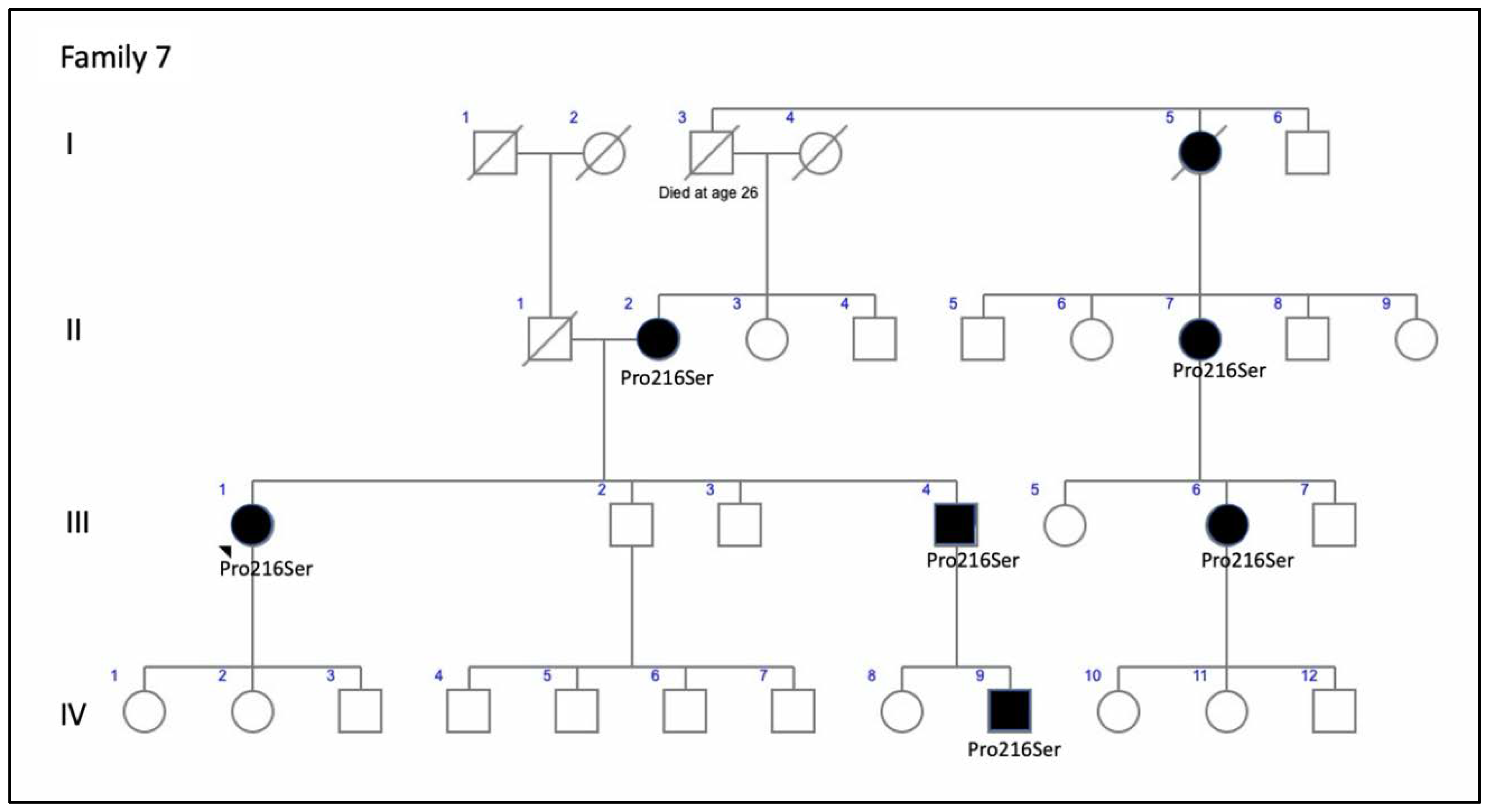
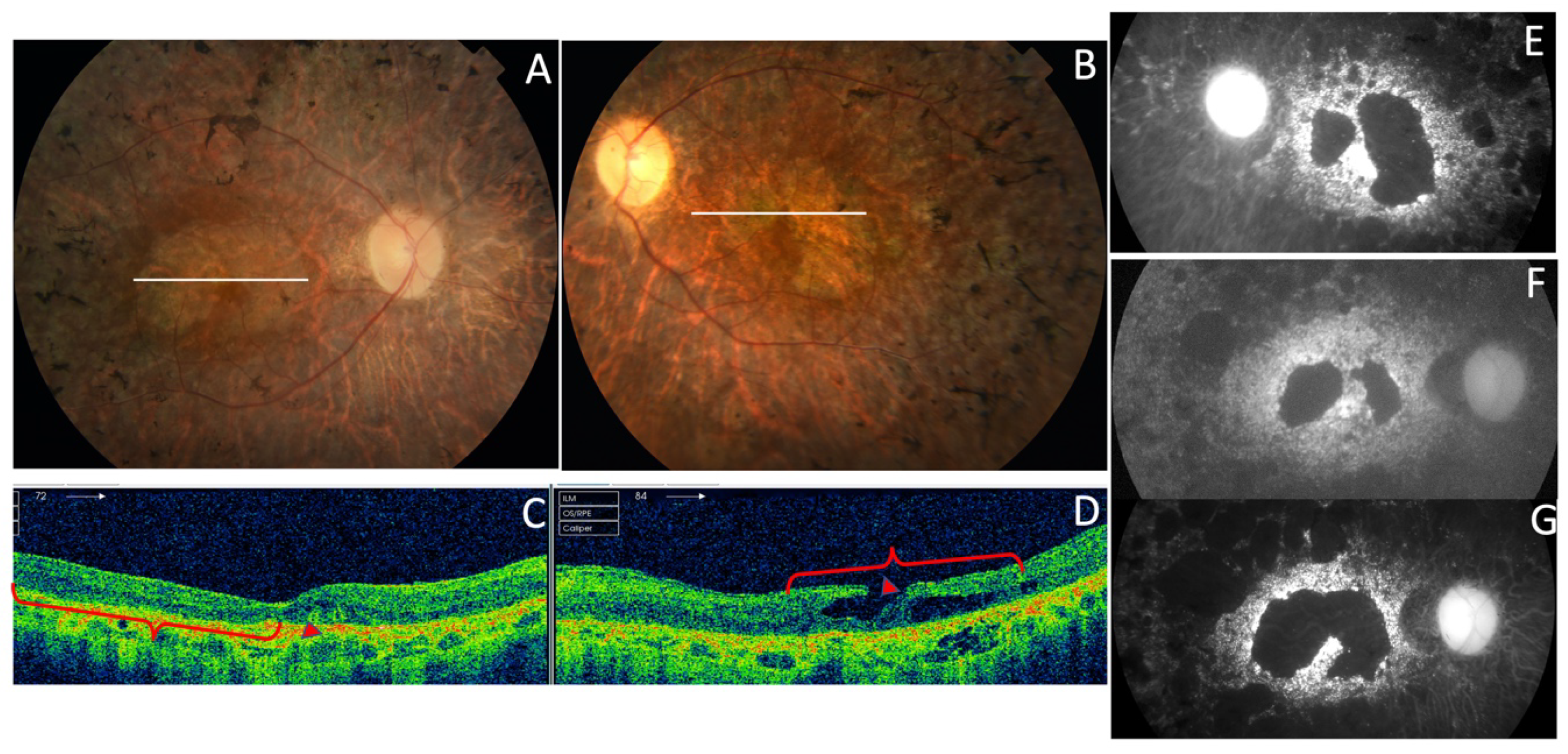
| F7-III-1 p.Pro216Ser | Fe | 18 | Tunnel vision and night blindness | 20/20 RE; 20/32 LE (48) | 20/25 RE; 20/200 LE (67) | Concentric retraction of visual field | Scotopic and photopic abolished |
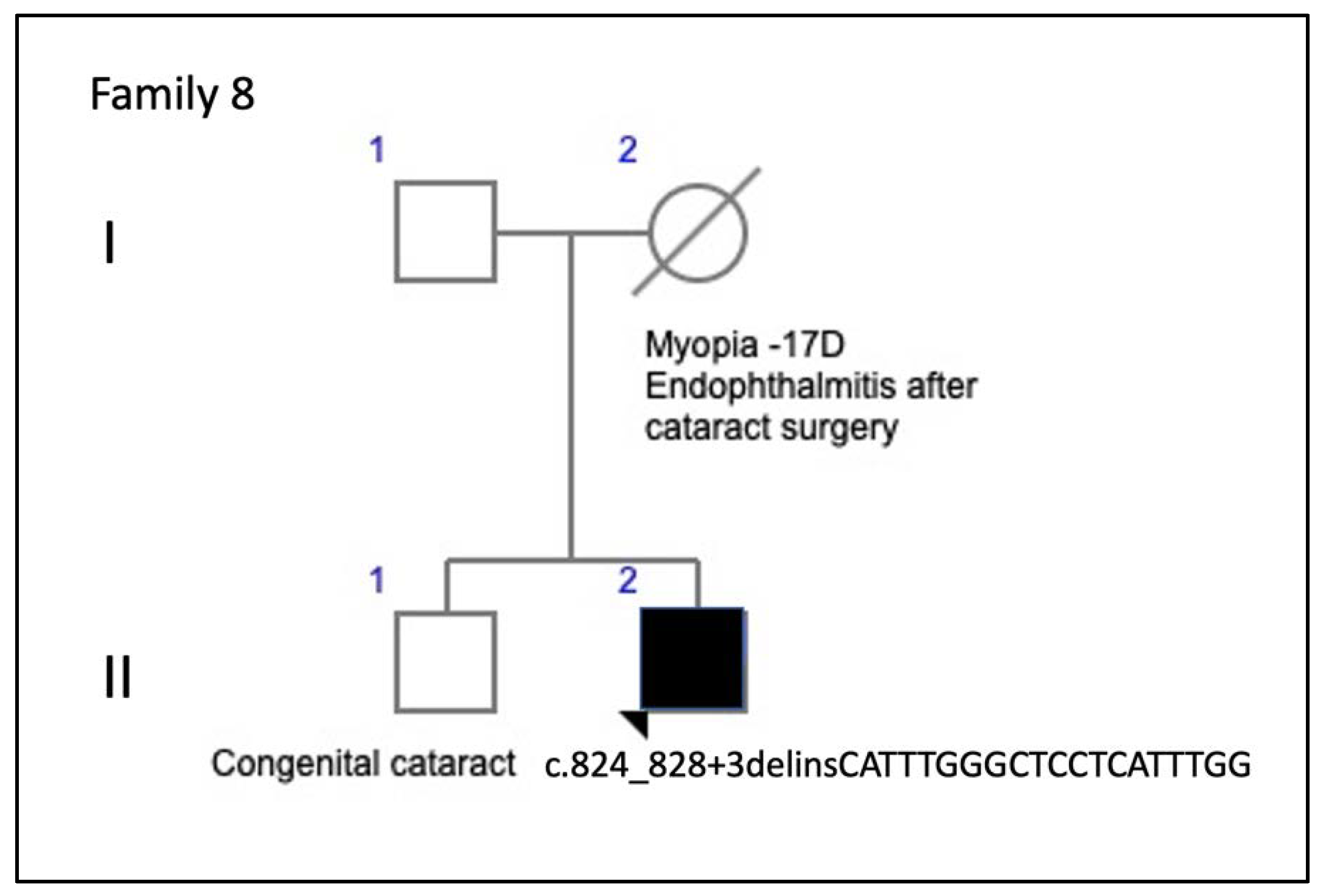
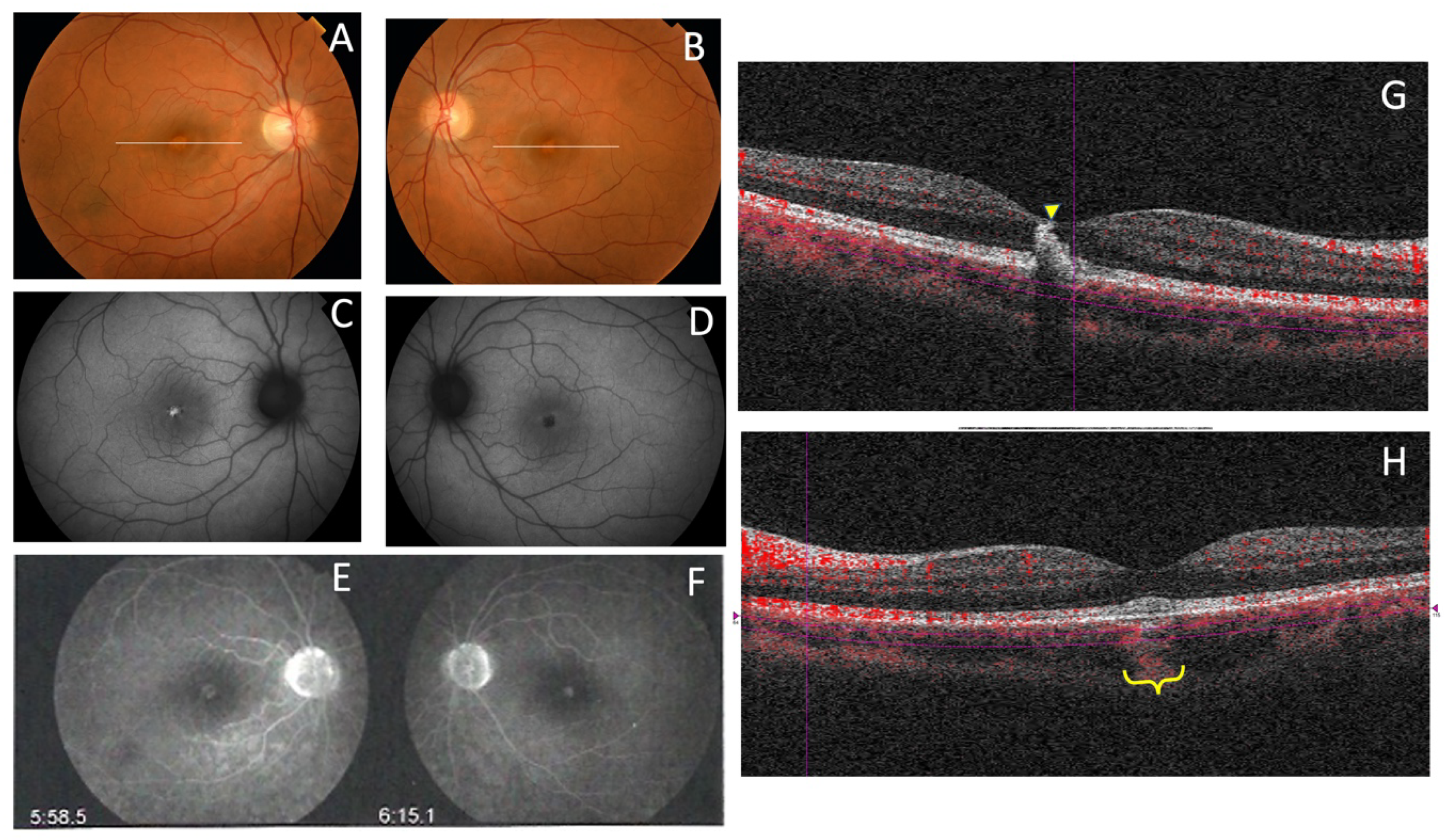
| F8-II-2 c.824_828+3delinsCATTTGGGCTCCTCATTTGG | M | 34 | Metamorphopsia | 20/25 RE; 20/40 LE (34) | N/A | Normal | Normal |
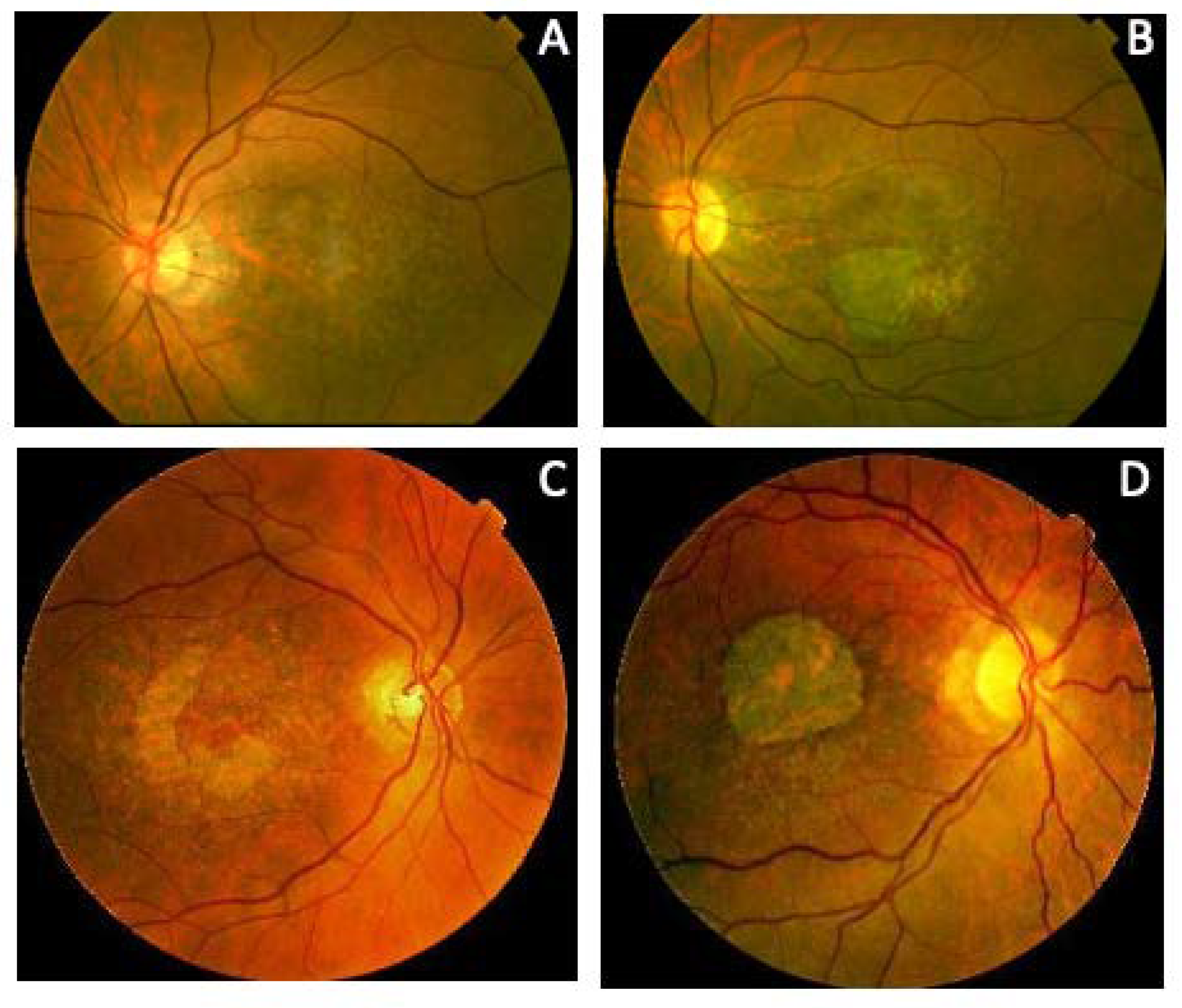
| Patient (Gene Variant) | Fundus at Central Retina | Fundus Rat Peripheral Retina | Autofluorescence | OCT | Clinical Diagnosis |
|---|---|---|---|---|---|
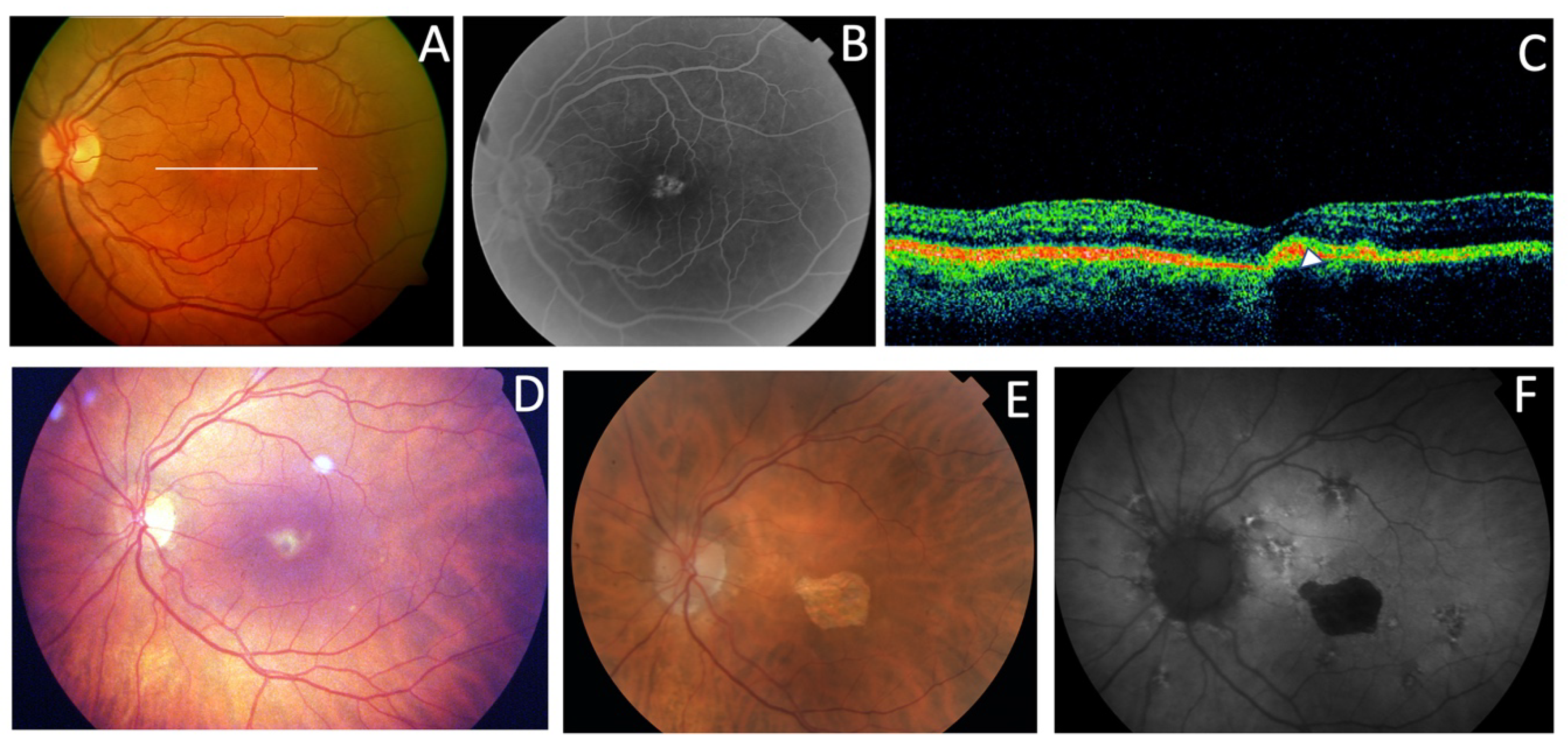
| F1-II-2 p.Arg46Ter | Subfoveal yellowish deposit | Normal | Hyper-AF of the subfoveal deposit | Hyper-reflective deposit above RPE in the foveal/perifoveal regions | AVMD |
| F1-II-3 p.Arg46Ter | Subfoveal yellowish deposit | Normal | Hyper-AF of the subfoveal deposit | Hyper-reflective deposit above the RPE in the foveal/perifoveal regions | AVMD |
| F1-II-4 p.Arg46Ter | Subfoveal yellowish deposit | Normal | Hyper-AF of the subfoveal deposit | Hyper-reflective deposit above the RPE in the foveal/perifoveal regions | AVMD |
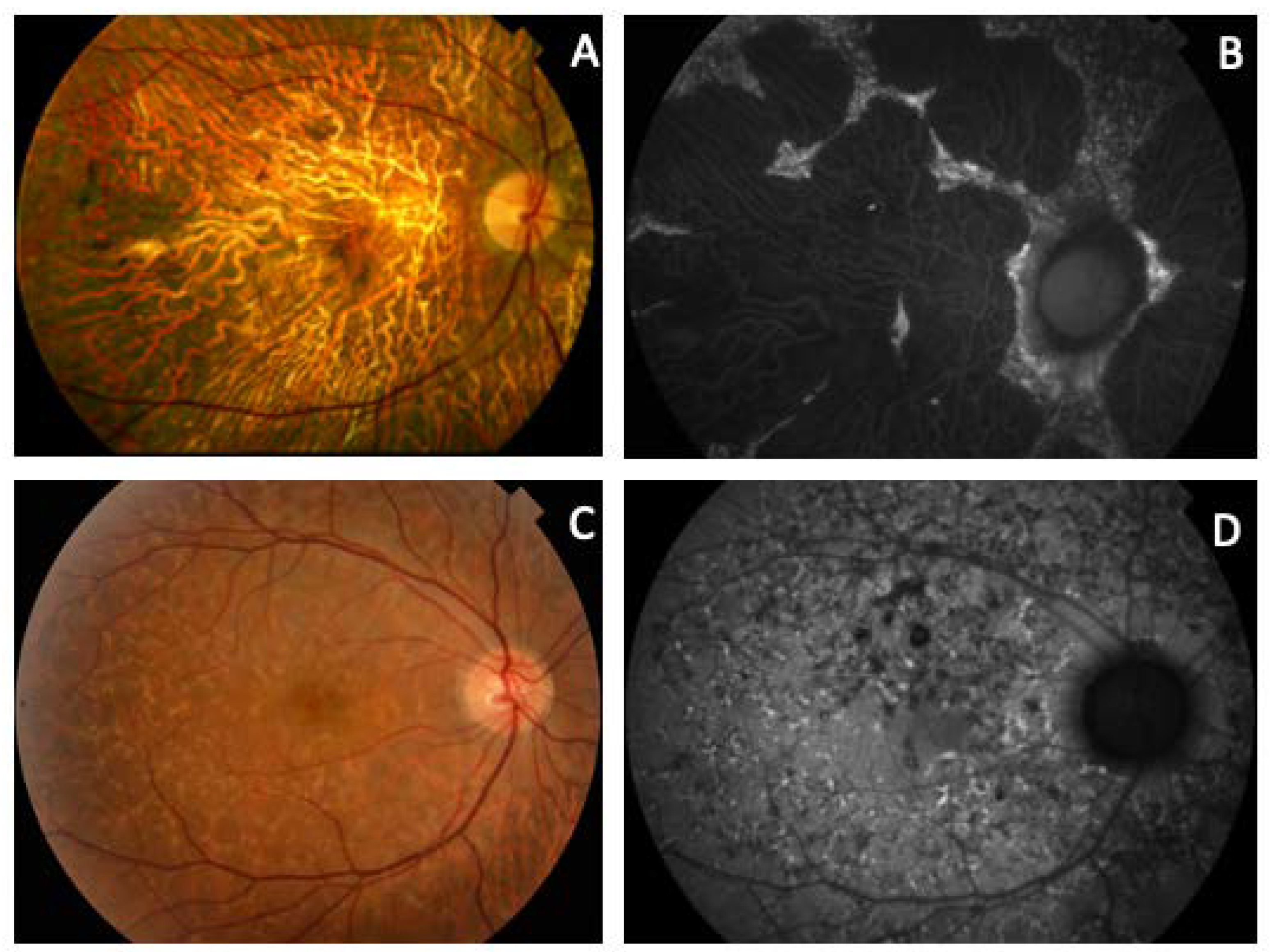
| F2-III-6 PRPH2: p.Arg46Ter ABCA4: p.Leu2027Phe/p.Gly1977Ser | Large confluent areas of CR atrophy, with preserved foveal area | Plaques of CR atrophy | Hypo-AF due to plaques atrophy in macula and periphery of the retina | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect at the posterior pole in BE. CRT = 128 μm RE/141 μm LE | Extensive CR atrophy |
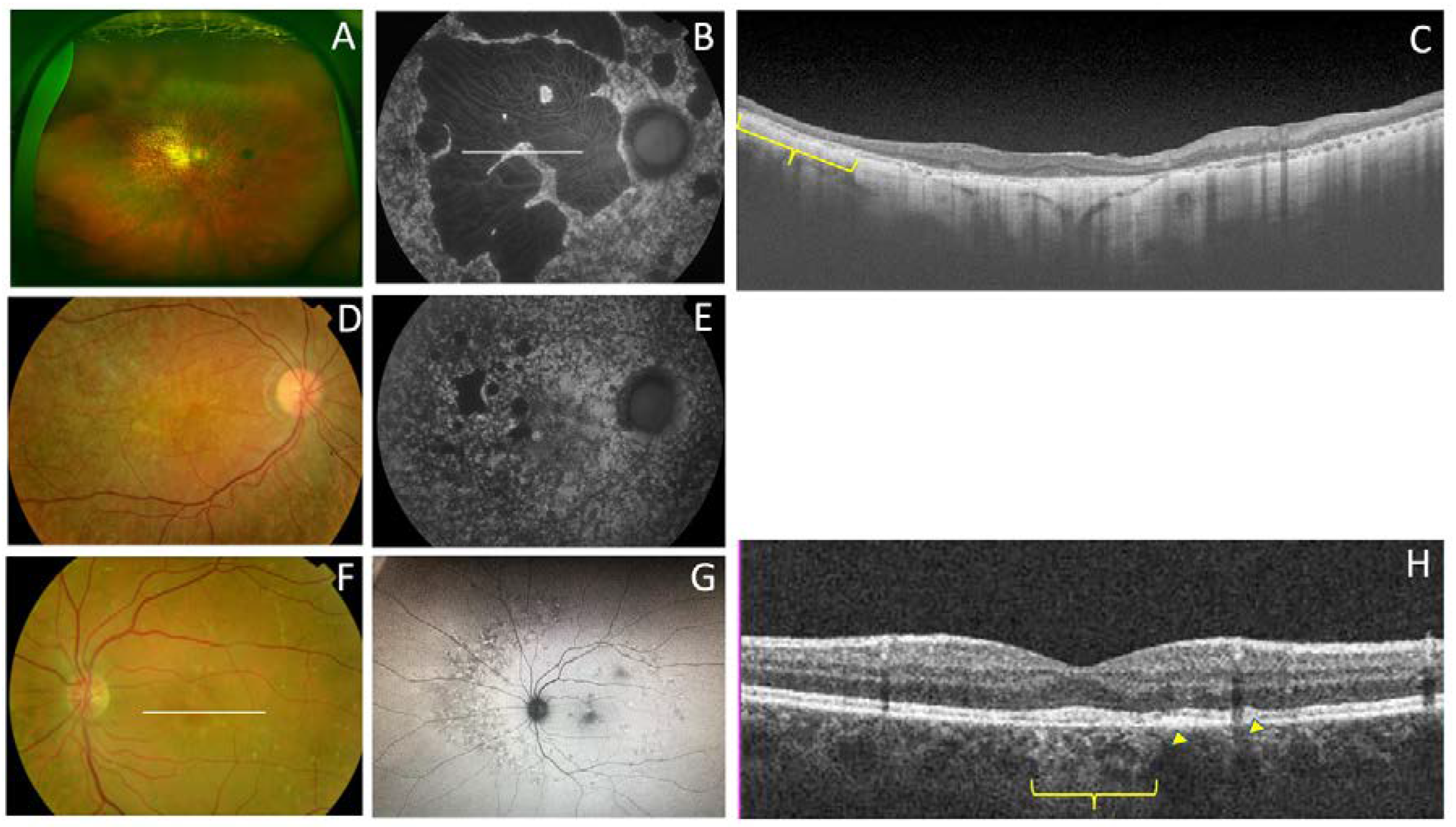
| F3-V-2 PRPH2: p.Lys154del ABCA4: p.Arg2030Gln | Yellow triradiate flecks in the posterior pole | Yellow and gray triradiate flecks in mid-periphery | Some flecks were hyper-AF and others were hypo-AF | Irregular aspect or disruption of the EZ and IZ limited to the foveal region. CRT = 286 μm RE/287 μm LE | Multifocal PD simulating Fundus Flavimaculatus |
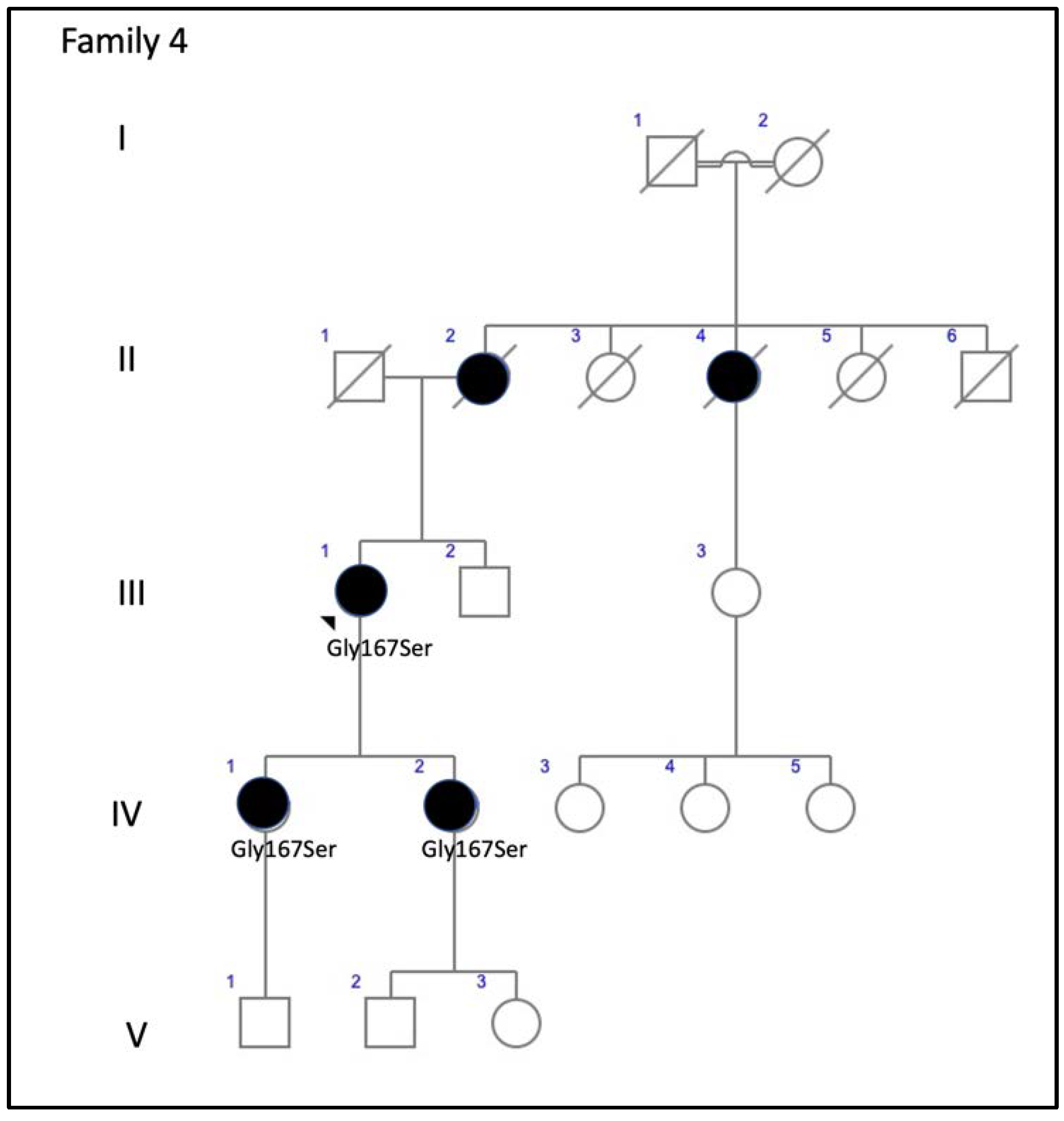
| F4-III-1 p.Gly167Ser | Large confluent areas of CR atrophy, with preserved foveal area | Whitish dots in mid-periphery | Confluent hypo-AF areas involving the optic disc and extending beyond the vascular arcades with speckled points of hypo-AF in the posterior pole and mid-periphery | Areas of outer retinal atrophy, EZ and IZ disrupted at the posterior pole in BE. Hyporeflective cysts at the inner nuclear layers and epiretinal membrane in the LE. CRT = 253 μm RE/252 μm LE | ADRP |
| F4-IV-1 p.Gly167Ser | Whitish stippling all over the posterior pole | Whitish dots in mid-periphery and small areas of atrophy in far-periphery | Small plaque area of hypo-AF at the fovea with scarce speckled points of hypo-AF and hyper-AF in the posterior pole and mid-periphery | EZ and IZ disrupted at the perifoveal level in BE. CRT = 270 μm RE/272 μm LE | Extensive CR atrophy |
| F4-IV-2 p.Gly167Ser | Yellow triradiate flecks in the posterior pole | Yellow triradiate flecks in mid-periphery | Macular hypo-AF with speckled hyper-AF and hypo-AF at the posterior pole and mid-periphery | Irregular aspect or disruption of the EZ and IZ limited to the foveal region. CRT = 264 μm RE/266 μm LE | Multifocal PD simulating Fundus Flavimaculatus |
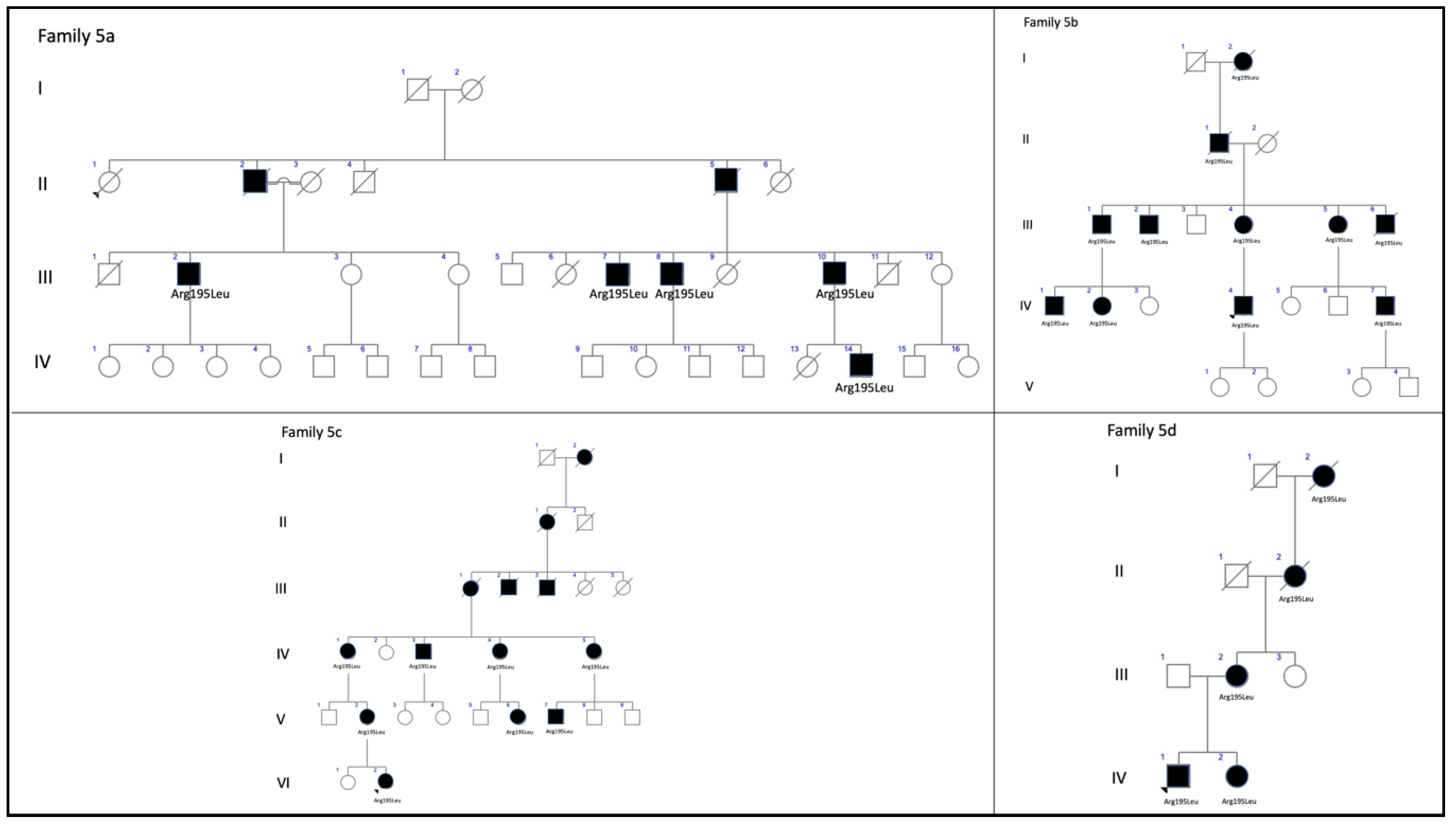
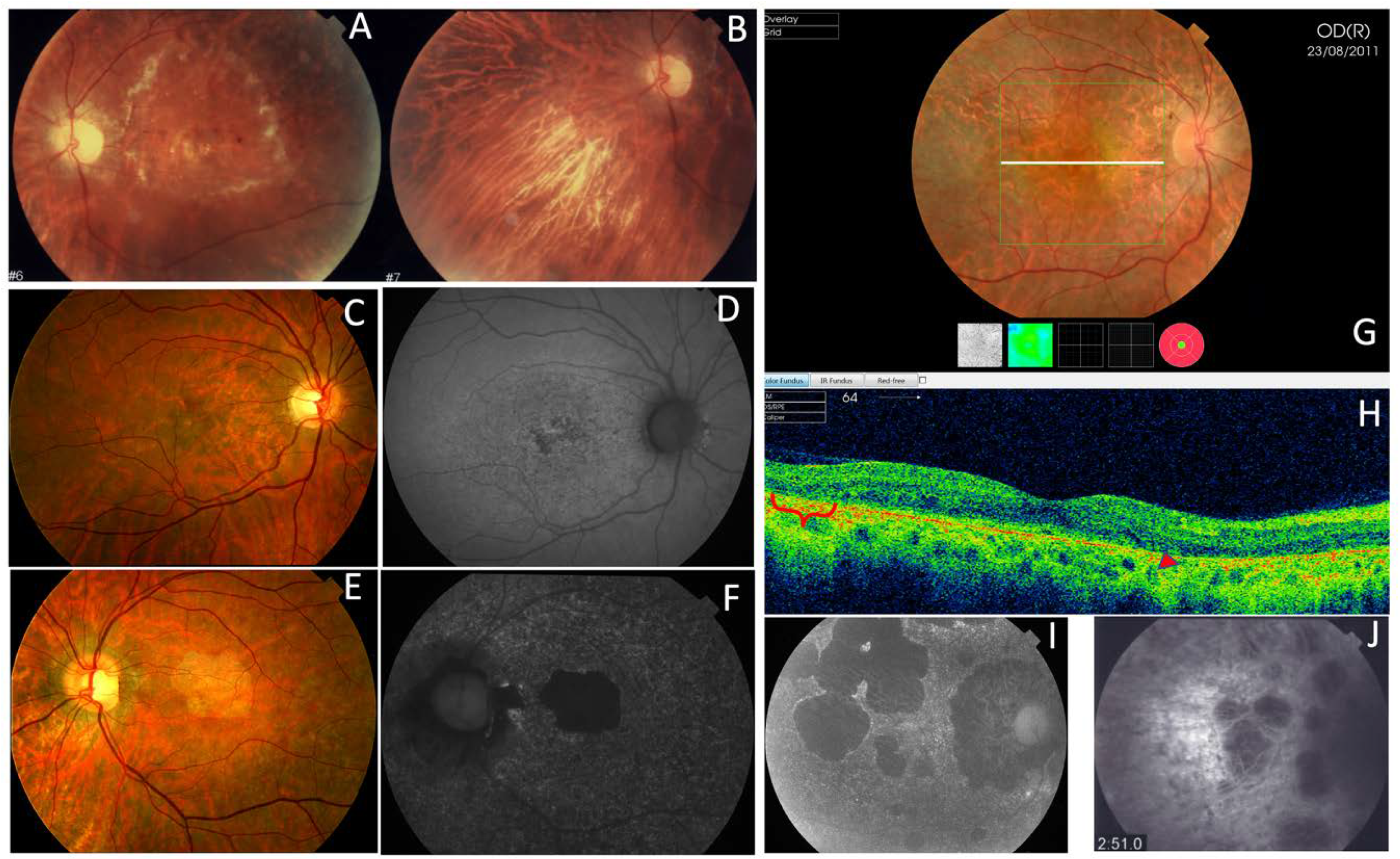
| F5a-III-2 p.Arg195Leu | Atrophy of both maculae with preserved fovea | Normal | Hypo-AF at the atrophic macular area and speckled hypo-AF within the vascular arcades | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect | CACD |
| F5a-III-7 p.Arg195Leu | Whitish stippling only in the macular area | Normal | Hypo-AF at the atrophic macular area | Irregular aspect or disruption of the EZ and IZ at the macular region | CACD |
| F5a-III-8 p.Arg195Leu | Atrophy of both maculae | Normal | Hypo-AF at the atrophic macular area | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect | CACD |
| F5a-III-10 p.Arg195Leu | Large areas of CR atrophy and whitish stippling within the vascular arcades | Normal | Hypo-AF at the atrophic macular area and speckled hypo-AF within the vascular arcades | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect | CACD |
| F5b-III-4 p.Arg195Leu | Large confluent areas of CR atrophy in the posterior pole and mid-periphery | Extensive CR atrophy of mid-periphery | Confluent hypo-AF areas involving the optic disc and extending beyond the vascular arcades with speckled points of hypo-AF in the posterior pole and mid-periphery | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect at the posterior pole in BE | Extensive CR atrophy |
| F5b-IV-1 p.Arg195Leu | Whitish stippling all over the posterior pole of BE with macular atrophy (>LE) | Whitish dots in mid-periphery | Small plaque areas of hypo-AF with scarce speckled points of hypo-AF in the posterior pole and mid-periphery | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect, EZ and IZ disrupted at the posterior pole in BE. CRT = 174 μm RE/184 μm LE | Extensive CR atrophy |
| F5b-IV-2 p.Arg195Leu | Atrophy of both maculae with preserved fovea | Normal | Speckled points of hypo-AF in the posterior pole | Retinal thinning of the macular region. CRT = 205 μm RE/213 μm LE | CACD |
| F5b-IV-4 p.Arg195Leu | Large confluent areas of CR atrophy, with preserved foveal area at the beginning | Plaques of CR atrophy in mid-periphery | Confluent hypo-AF areas involving the optic disc and extending beyond the vascular arcades with speckled points of hypo-AF in the posterior pole and mid-periphery | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect, EZ and IZ disrupted at the posterior pole in BE. CRT = 220 μm RE/214 μm LE | Extensive CR atrophy |
| F5c-IV-4 p.Arg195Leu | Atrophy of both maculae | Small areas of CR atrophy in the RE | Small plaque areas of hypo-AF in the posterior pole BE and in the mid-periphery RE | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect. CRT = 179 μm RE/191 μm LE | CACD |
| F5c-V-2 p.Arg195Leu | Very small, whitish dots all over the posterior pole | Very small, shining, whitish dots in mid-periphery | Speckled points of hypo-AF in the posterior pole and mid-periphery | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect, EZ and IZ disrupted at the posterior pole in BE | Extensive CR atrophy |
| F5c-VI-2 p.Arg195Leu | Very small, whitish dots all over the posterior pole | Very small, shining, whitish dots in mid-periphery | Speckled points of hypo-AF in the posterior pole and mid-periphery | Retinal thinning of the macular region | Extensive CR atrophy |
| F5d-VI-1 p.Arg195Leu | Large confluent areas of CR atrophy, with preserved foveal area at the beginning | Small areas of CR atrophy in BE | Large plaque areas of hypo-AF in the posterior pole and small plaque areas in mid-periphery | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect at the posterior pole in BE | ADRP |
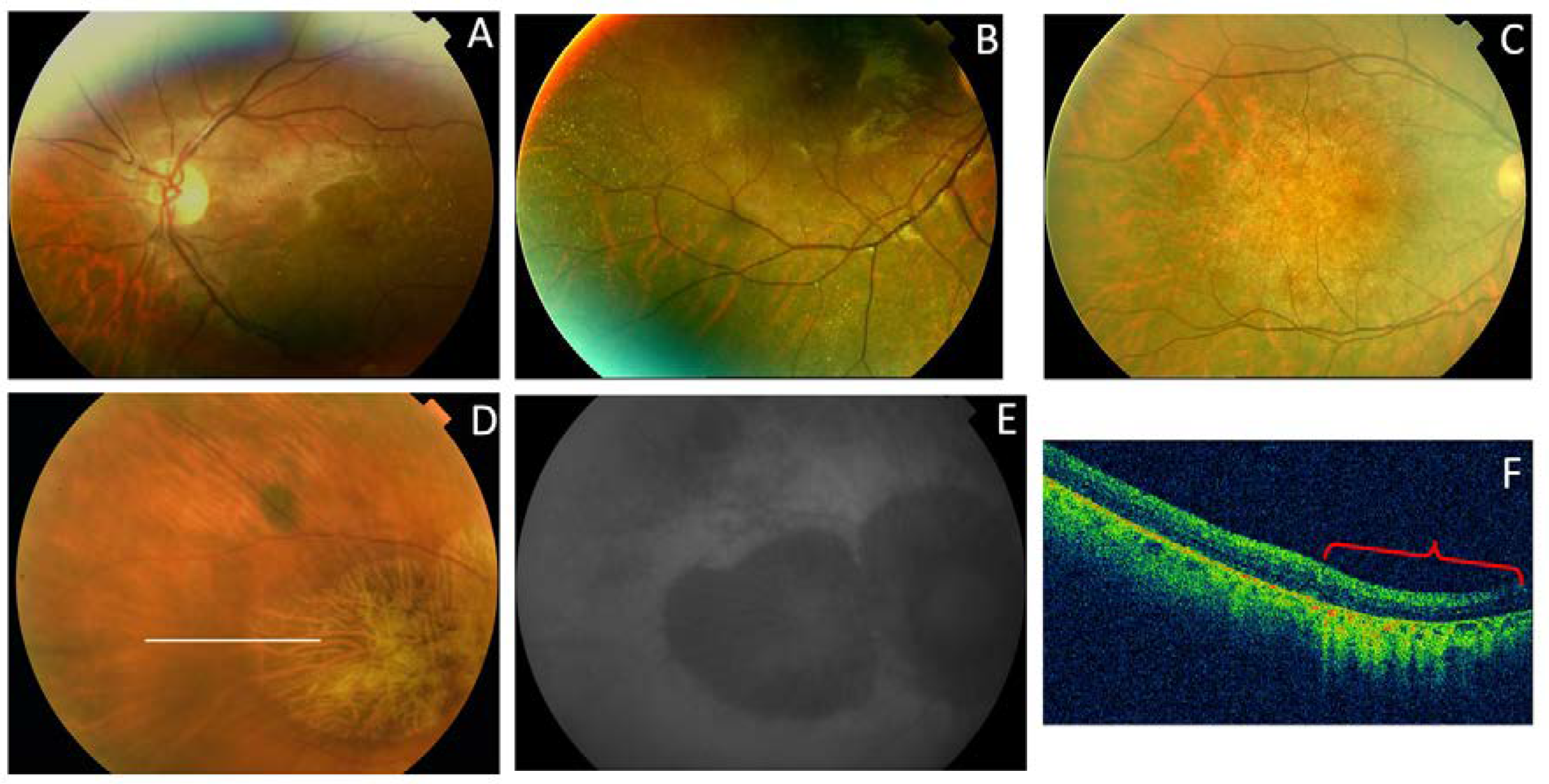
| F6-I-5 p.Val209Ile | Subfoveal yellowish deposit that evolved to macular atrophy | Normal | Hyper-AF first, followed by macular hypo-AF | Hyper-reflective deposit above the RPE in the foveal region followed by outer retinal atrophy of the macular region. CRT = 179 μm RE/164 μm LE | AVMD |
| F6-II-2 p.Val209Ile | Drusen-like deposits nasal to fovea in the LE | Normal | Juxtafoveal hypo-AF spots | Small scarce hyper-reflective deposit above the RPE in the perifoveal region. CRT = 226 μm RE/226 μm LE | AVMD |
| F7-III-1 p.Pro216Ser | CR atrophy (>LE) | CR atrophy and pigmentation in spicules | Hypo-AF in macula and periphery of the retina | Outer retinal atrophy of the macular region and choroidal hyper-reflectivity by window defect at the posterior pole in BE. Hyporeflective cysts at the inner nuclear layer and lamellar macular hole in the LE. CRT = 188 μm RE/258 μm LE | ADRP |
| F8-II-2 c.824_828+3delinsCATTTGGGCTCCTCATTTGG | Subfoveal yellowish deposit | Normal | Hyper-AF deposit in RE and hypo-AF in LE | Hyper-reflective deposit above the RPE in the foveal/perifoveal regions | AVMD |
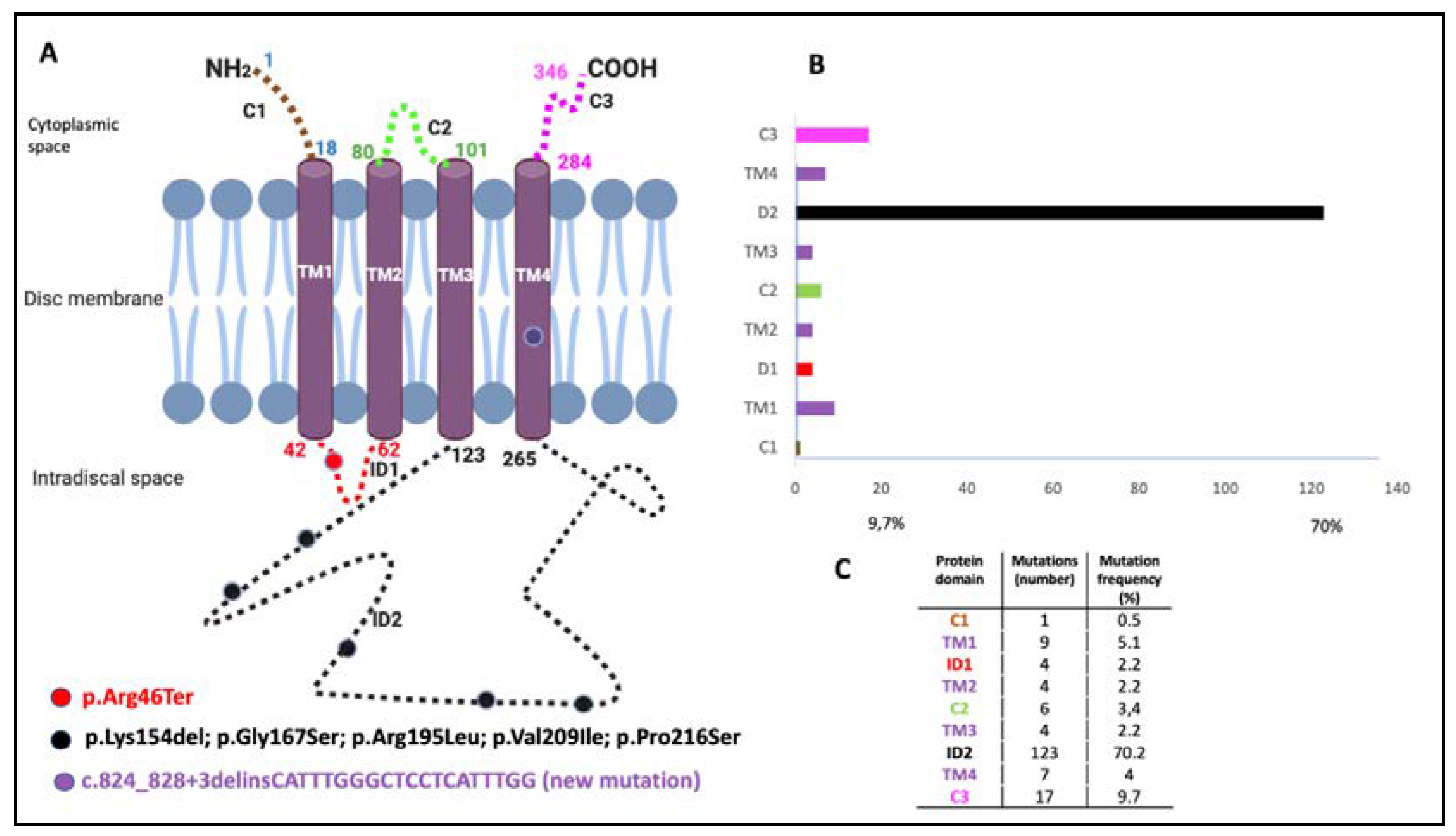
| Family | PRPH2 Gene Mutations | PRPH2 Gene Mutation Type | Location of Mutation in the Prph2 Protein Domain | Global Allele Frequency | Accession Number | Phenotypes of our Patients | All Phenotypes Described |
|---|---|---|---|---|---|---|---|
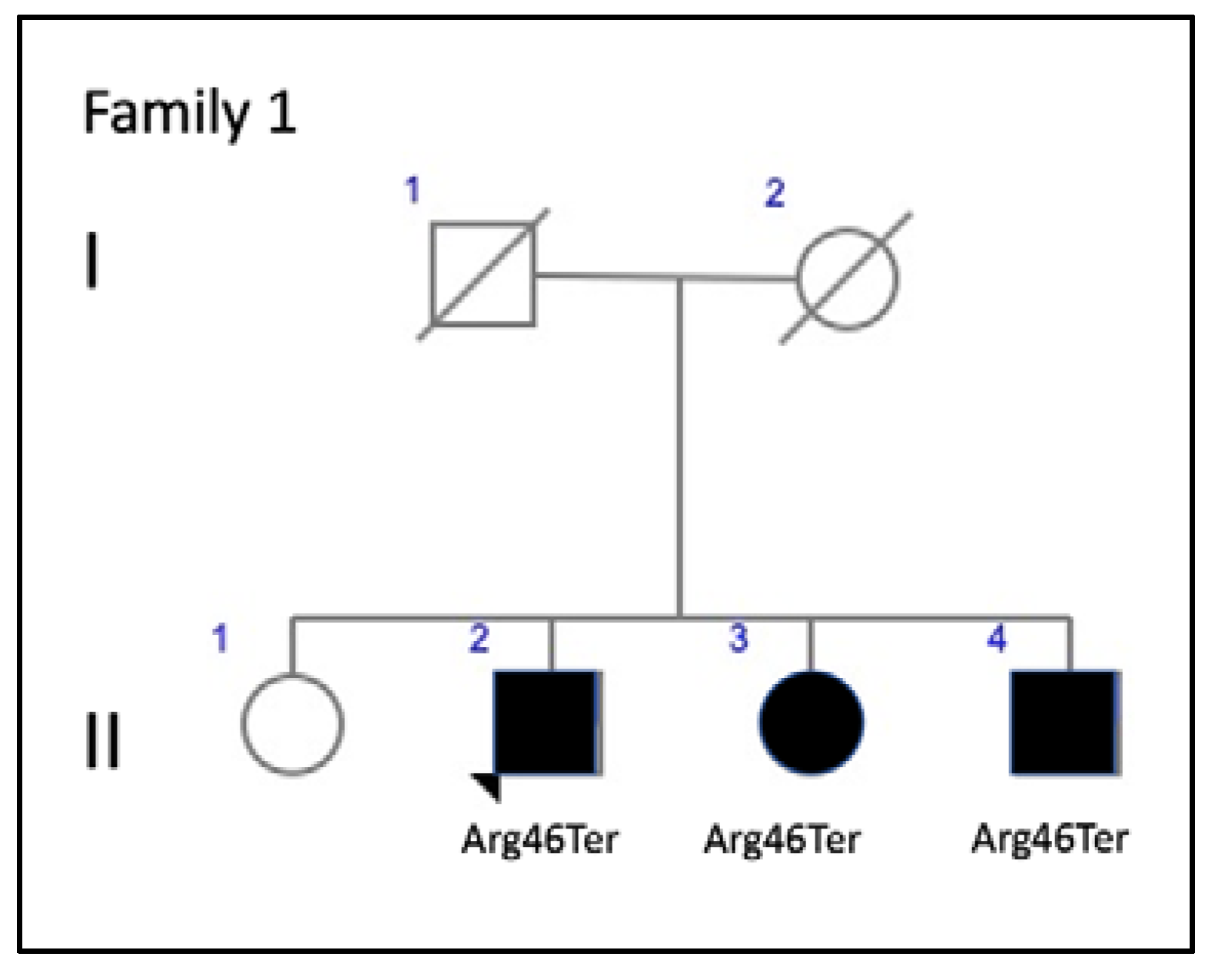
| 1 (3 patients) | c.136C>T, p.Arg46Ter | Nonsense | ID1 | 1.59115 × 10−5 | rs61755771 | AVMD: F1-II-2, F1-II-3, and F1-II-4 | AVMD [13] |
| 2 (1 patient) | c.136C>T, p.Arg46Ter +ABCA4: c.6079C>T, p.Leu2027Phe and c.5929G>A, p.Gly1977Ser | Nonsense | ID1 | 1.59115 × 10−5 | rs61751408 rs61750639 | ECA: F2-III-6 | ECA [current study] (blended phenotype) |
| 3 (1 patient) | c.461_463del, p.Lys154del +ABCA4: c.6089G>A, p.Arg2030Gln | Amino acid deletion | ID2 | Unknown | rs61755786 rs61750641 | PDsFF: F3-V-2 | ADRP [14] PDsFF [current study] |
| 4 (3 patients) | c.499G>A, p.Gly167Ser | Missense | ID2 | 1.59280 × 10−5 | rs527236098 | ADRP: F4-III-1 ECA: F4-IV-1 PDsFF: F4-IV-2 | PD [15] PDsFF [current study] ECA [current study] ADRP [current study] |
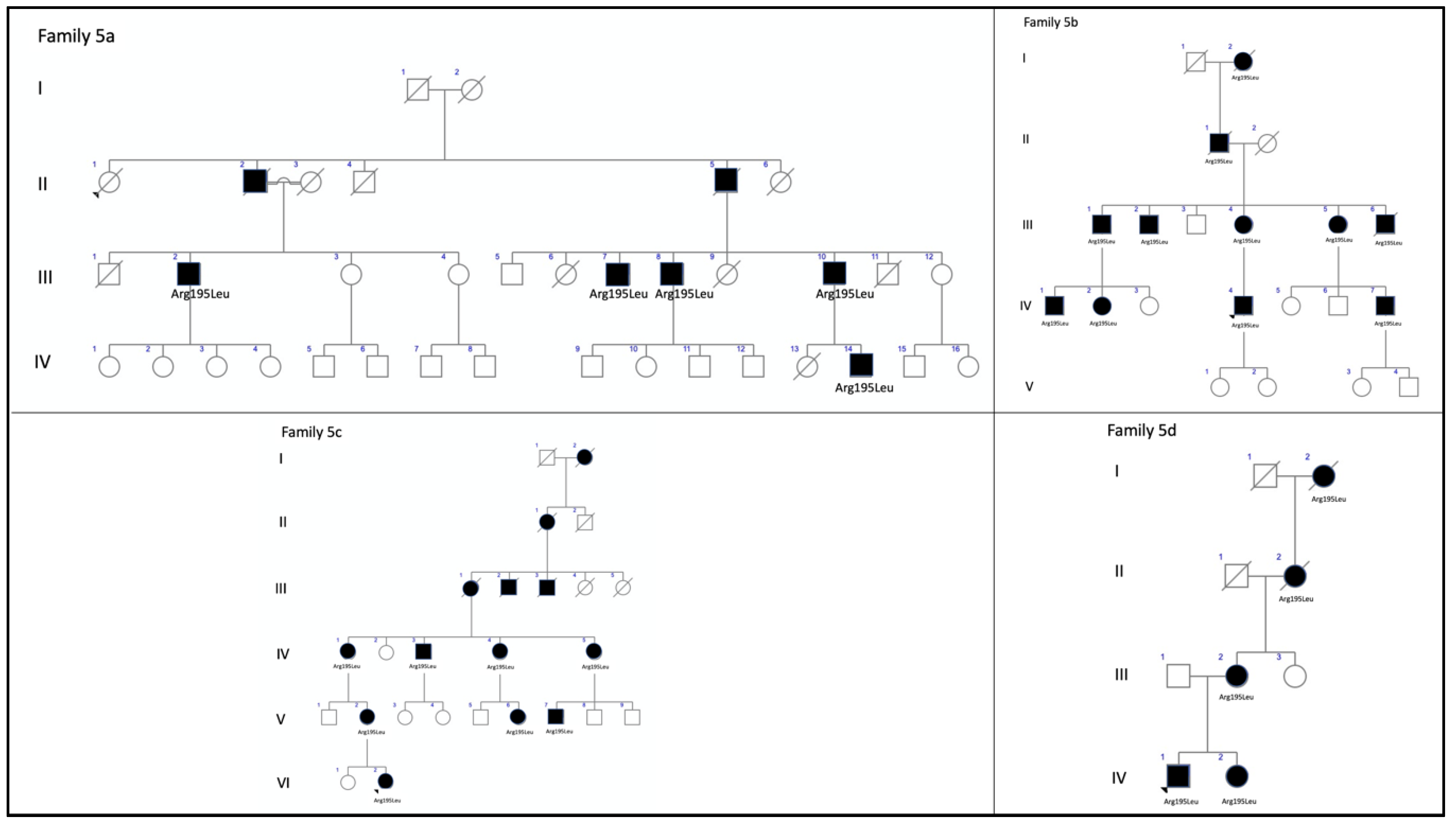
| 5 (12 patients) | c.584G>T, p.Arg195Leu | Missense | ID2 | 3.98349 × 10−6 | rs121918567 | CACD: F5a-III-2, F5a-III-7, F5a-III-8, F5aIII-10, F5b-IV-2, and F5c-IV-4 ECA: F5b-III-4, F5b_IV-1, F5b-IV-4, F5c-V-2, and F5c-VI-2 ADRP: F5d-VI-1 | CACD [16] ECA [13] ADRP [current study] |
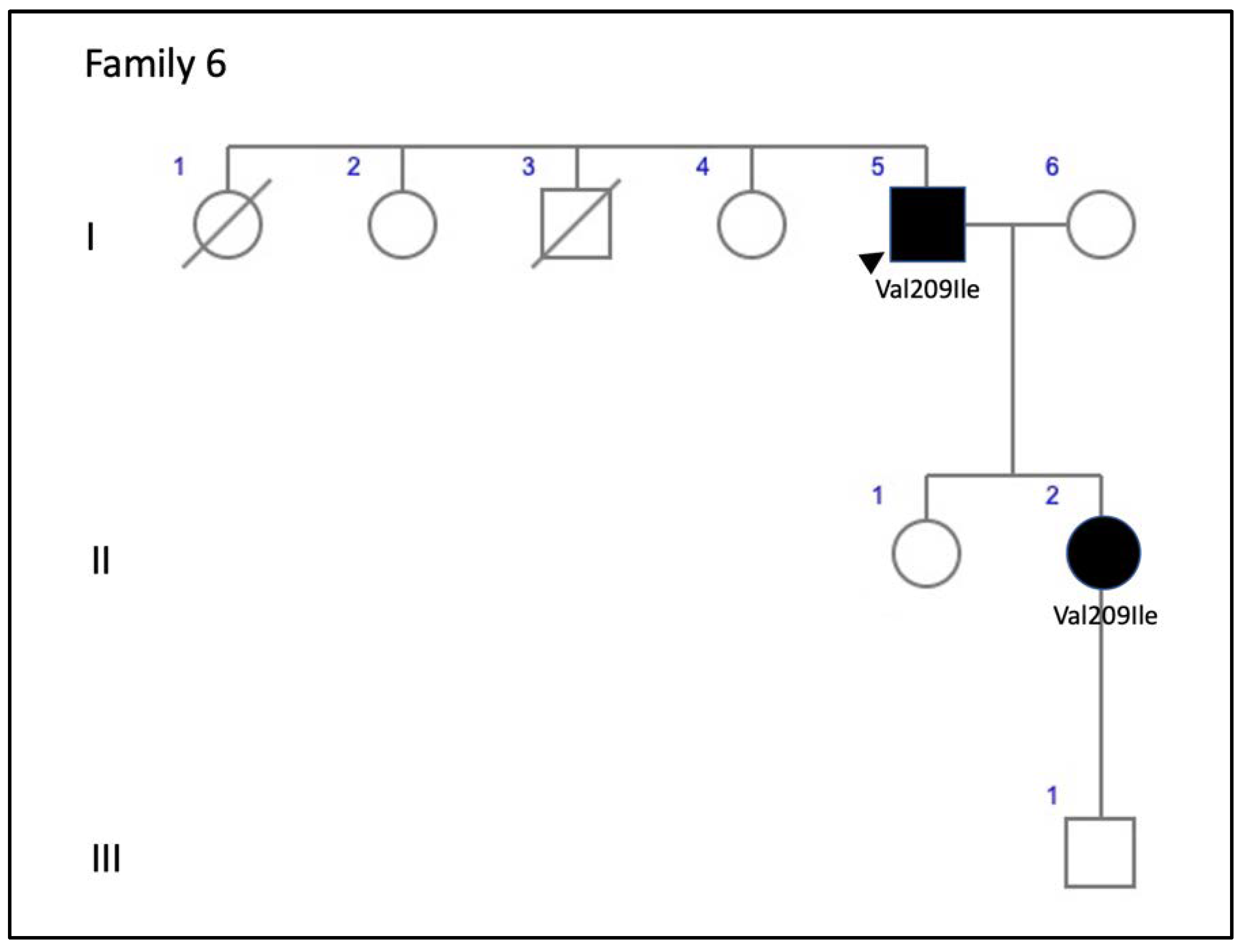
| 6 (2 patients) | c.625G>A, p.Val209Ile | Missense | ID2 | 1.98877 × 10−5 | rs753657349 | AVMD: F6-I-5 and F6-II-2 | AVMD [13] |
| 7 (1 patient) | c.646C>T, p.Pro216Ser | del | ID2 | Unknown | rs61755805 | ADRP: F7-III-1 | ADRP [17] |
| 8 (1 patient) | c.824_828+3delinsCATTTGGGCTCCTCATTTGG | del/ins | TM4 | Not previously described | AVMD: F8-II-2 | AVMD [current study] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coco-Martin, R.M.; Sanchez-Tocino, H.T.; Desco, C.; Usategui-Martín, R.; Tellería, J.J. PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes 2020, 11, 773. https://doi.org/10.3390/genes11070773
Coco-Martin RM, Sanchez-Tocino HT, Desco C, Usategui-Martín R, Tellería JJ. PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes. 2020; 11(7):773. https://doi.org/10.3390/genes11070773
Chicago/Turabian StyleCoco-Martin, Rosa M., Hortensia T. Sanchez-Tocino, Carmen Desco, Ricardo Usategui-Martín, and Juan J. Tellería. 2020. "PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation" Genes 11, no. 7: 773. https://doi.org/10.3390/genes11070773
APA StyleCoco-Martin, R. M., Sanchez-Tocino, H. T., Desco, C., Usategui-Martín, R., & Tellería, J. J. (2020). PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes, 11(7), 773. https://doi.org/10.3390/genes11070773