Computational Analysis of the Global Effects of Ly6E in the Immune Response to Coronavirus Infection Using Gene Networks

 ,
,  ,
,  ,
,  and
and 
Abstract
:
1. Introduction
2. Related Works
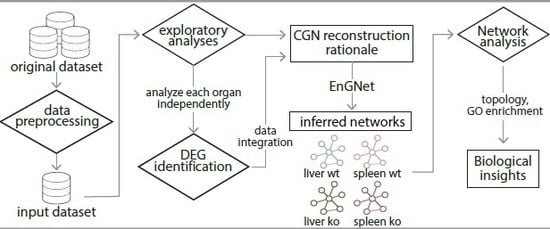
3. Materials and Methods
3.1. Original Dataset Description
3.2. Data Pre-Processing
3.3. Differential Expression Analyses
3.4. Inference of the Gene Networks: EnGNet
3.5. Networks Analyses
4. Results
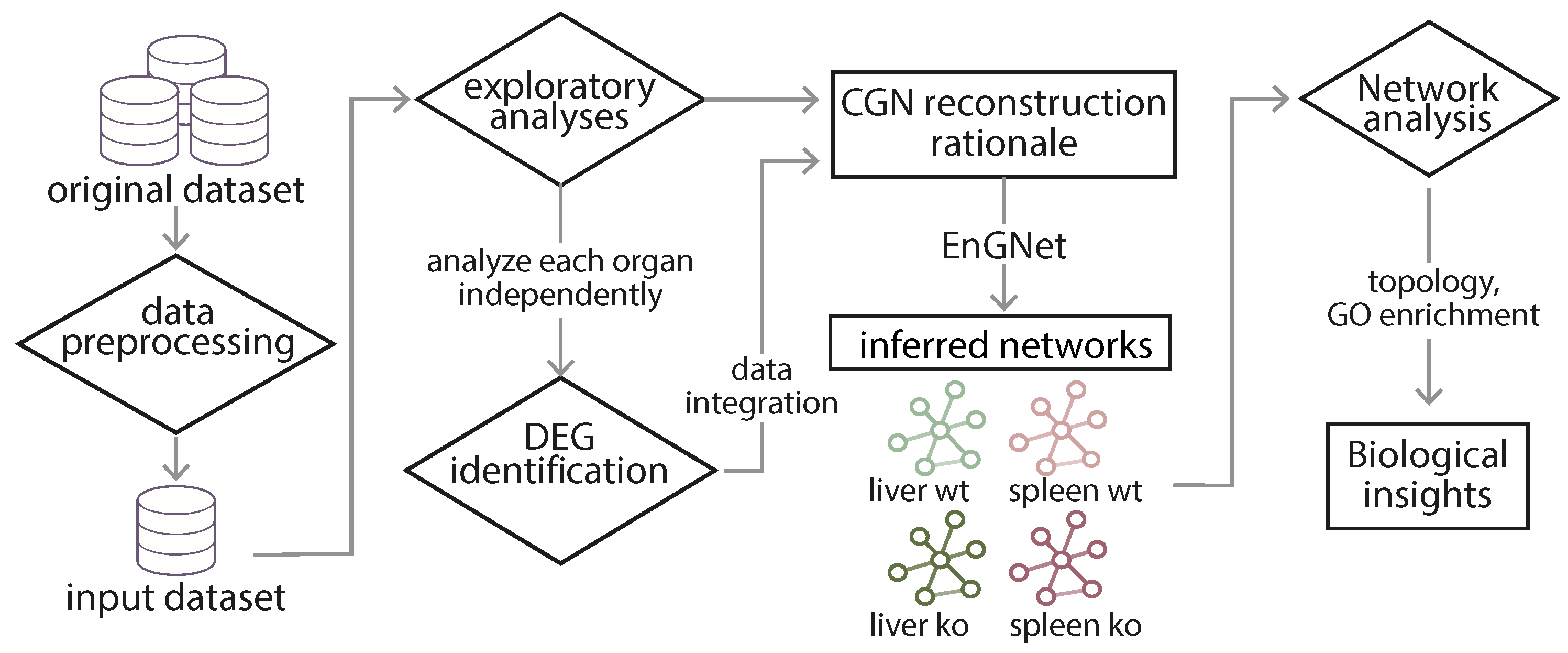
4.1. Data Pre-Processing and Exploratory Analyses
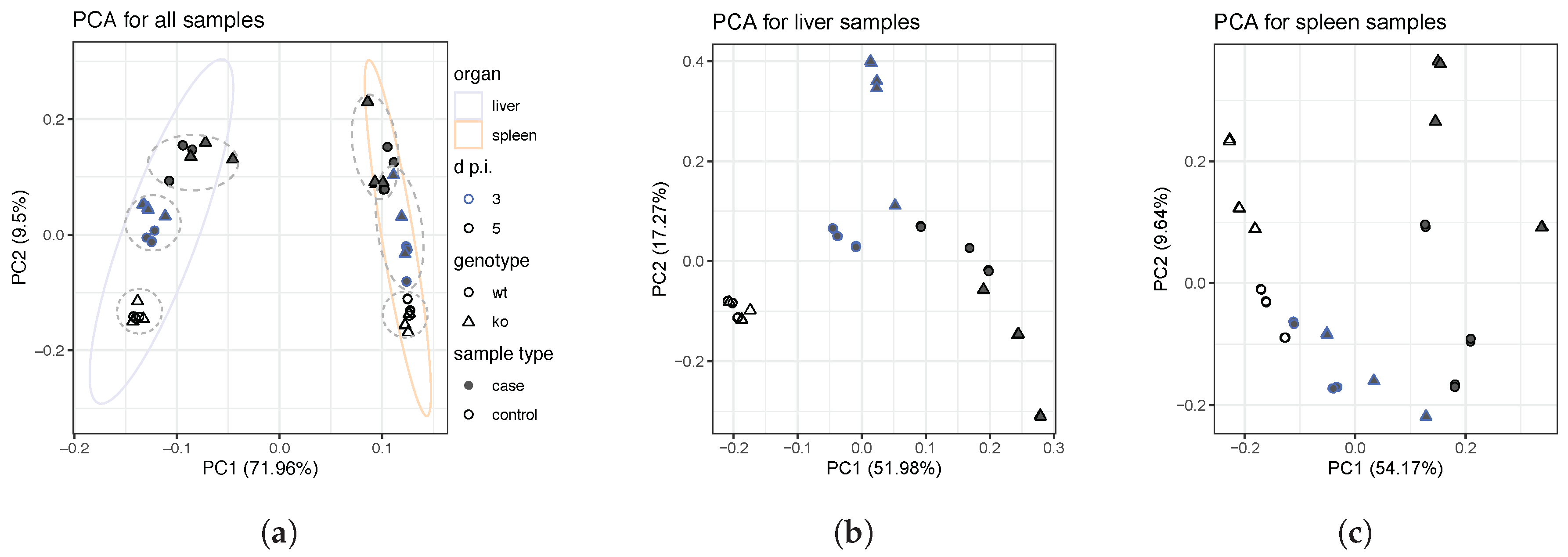
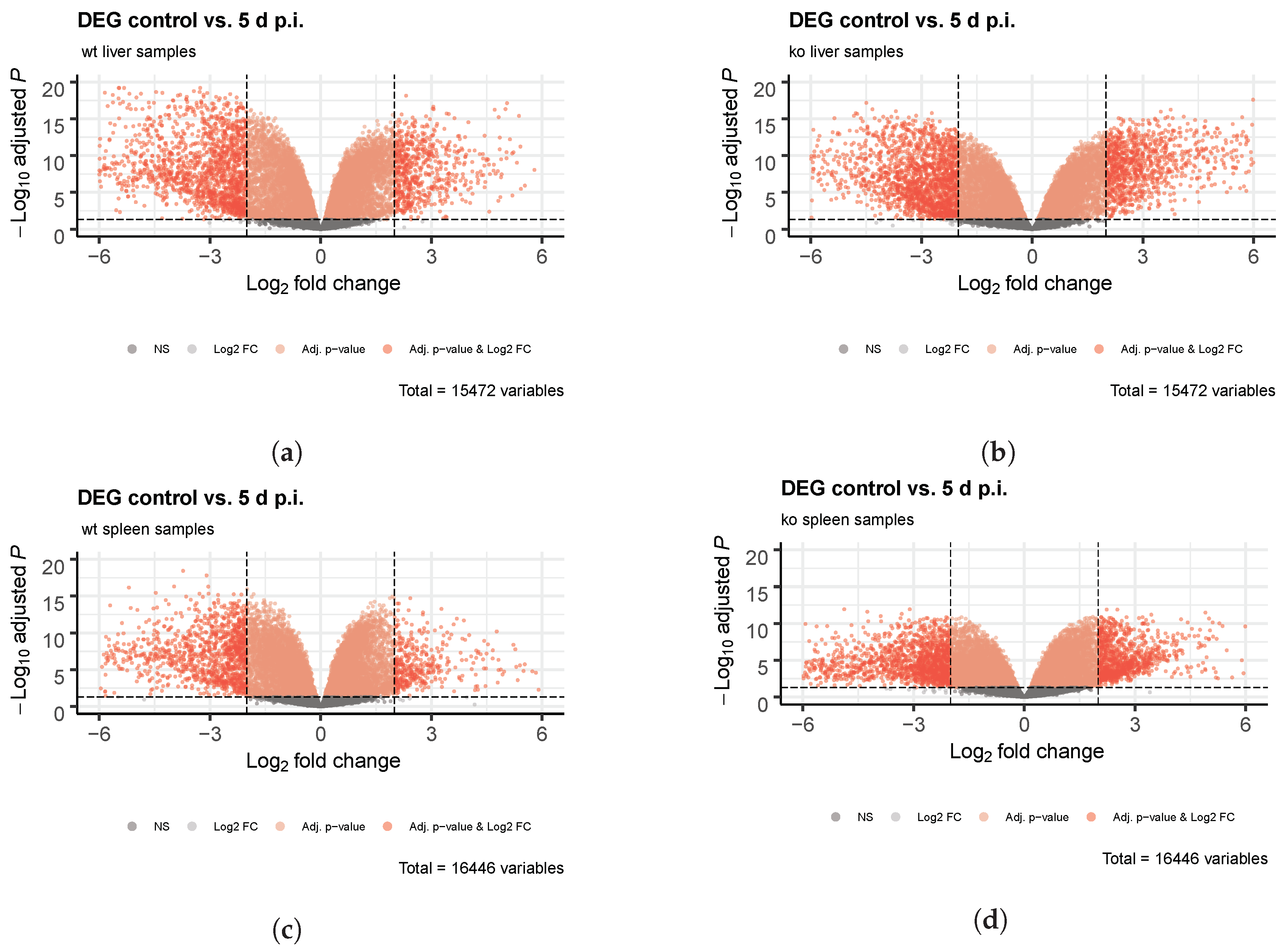
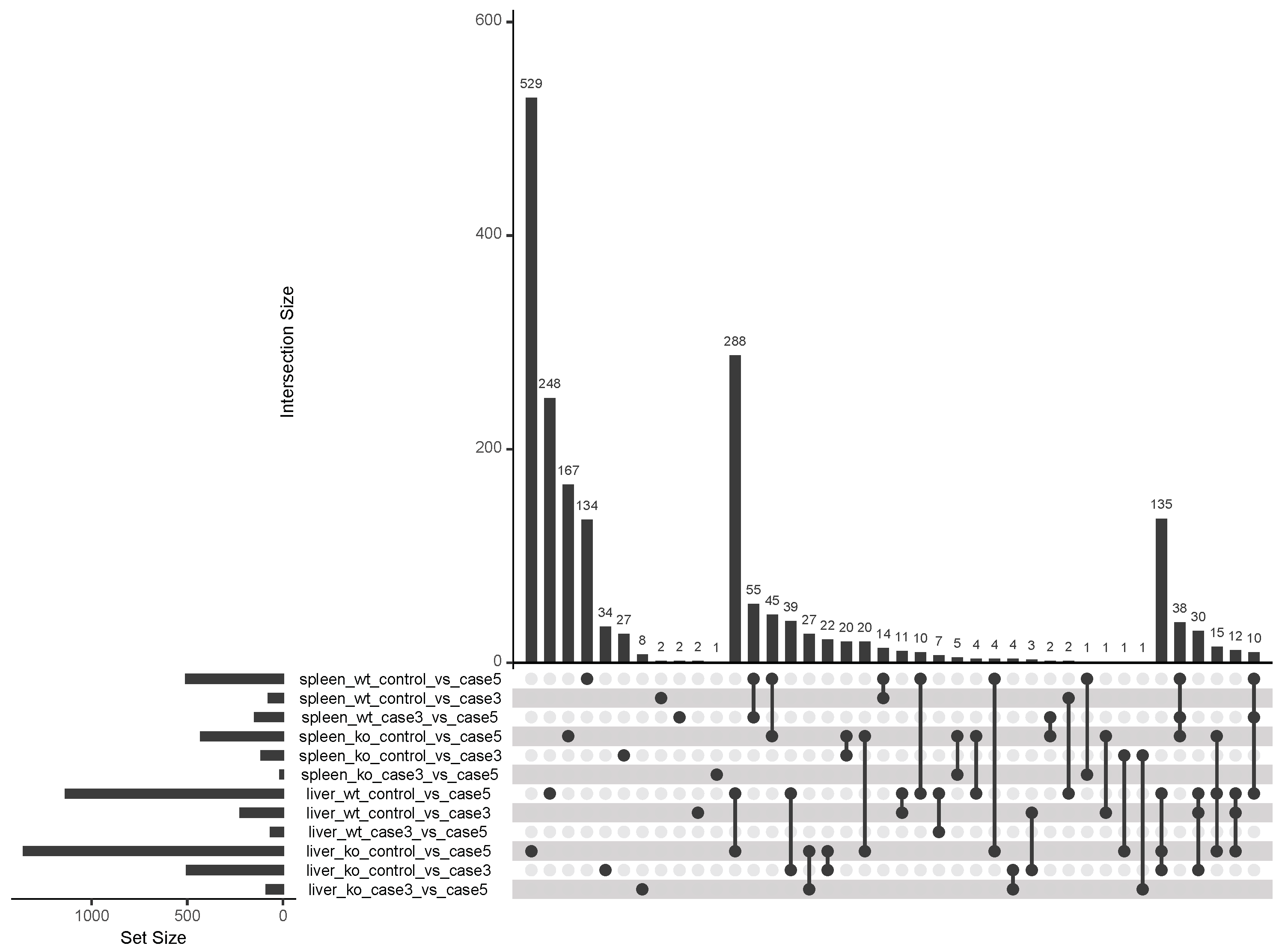
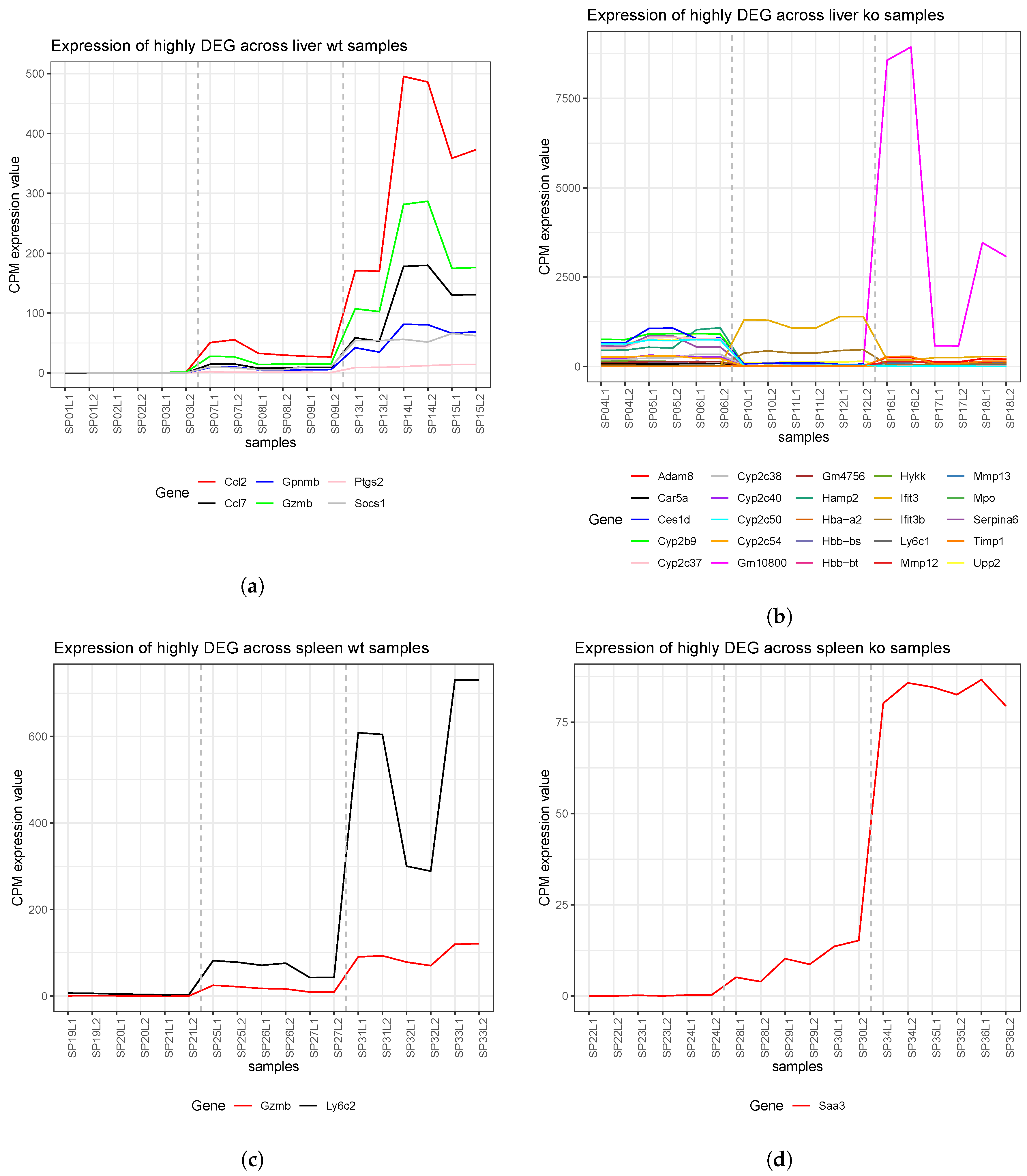
4.2. Identification of Differentially-Expressed Genes Between Wild Type and Ly6E Samples
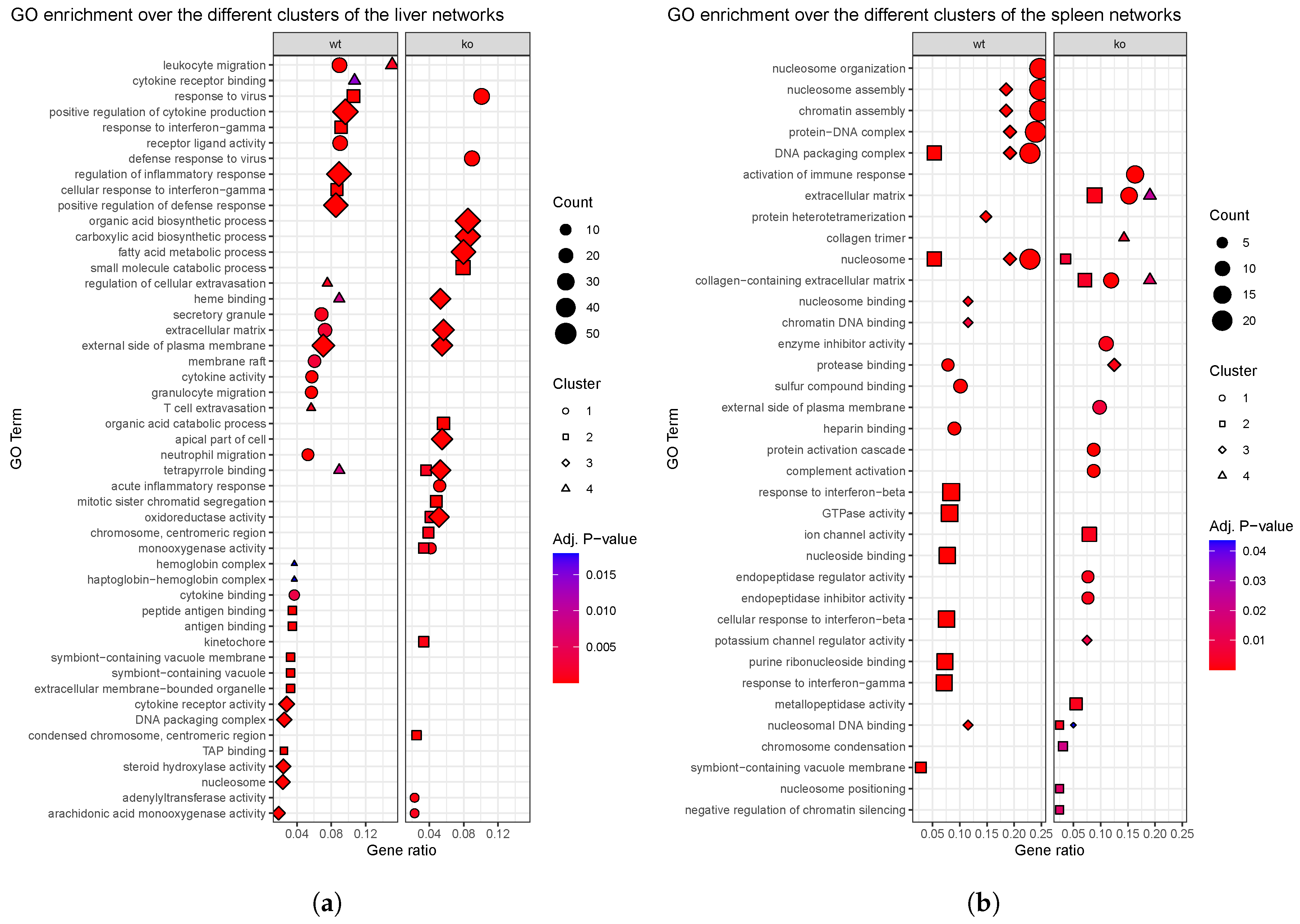
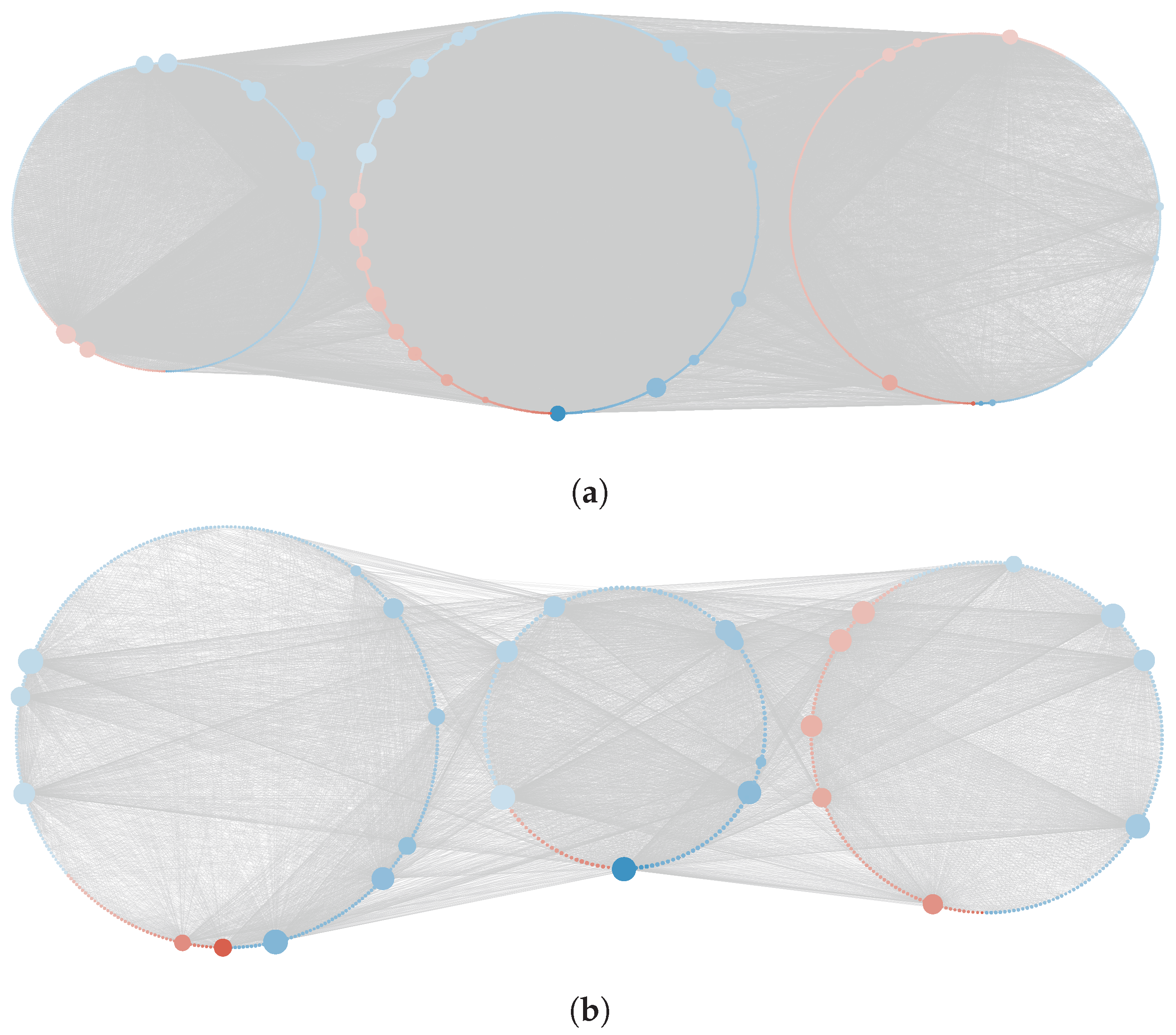
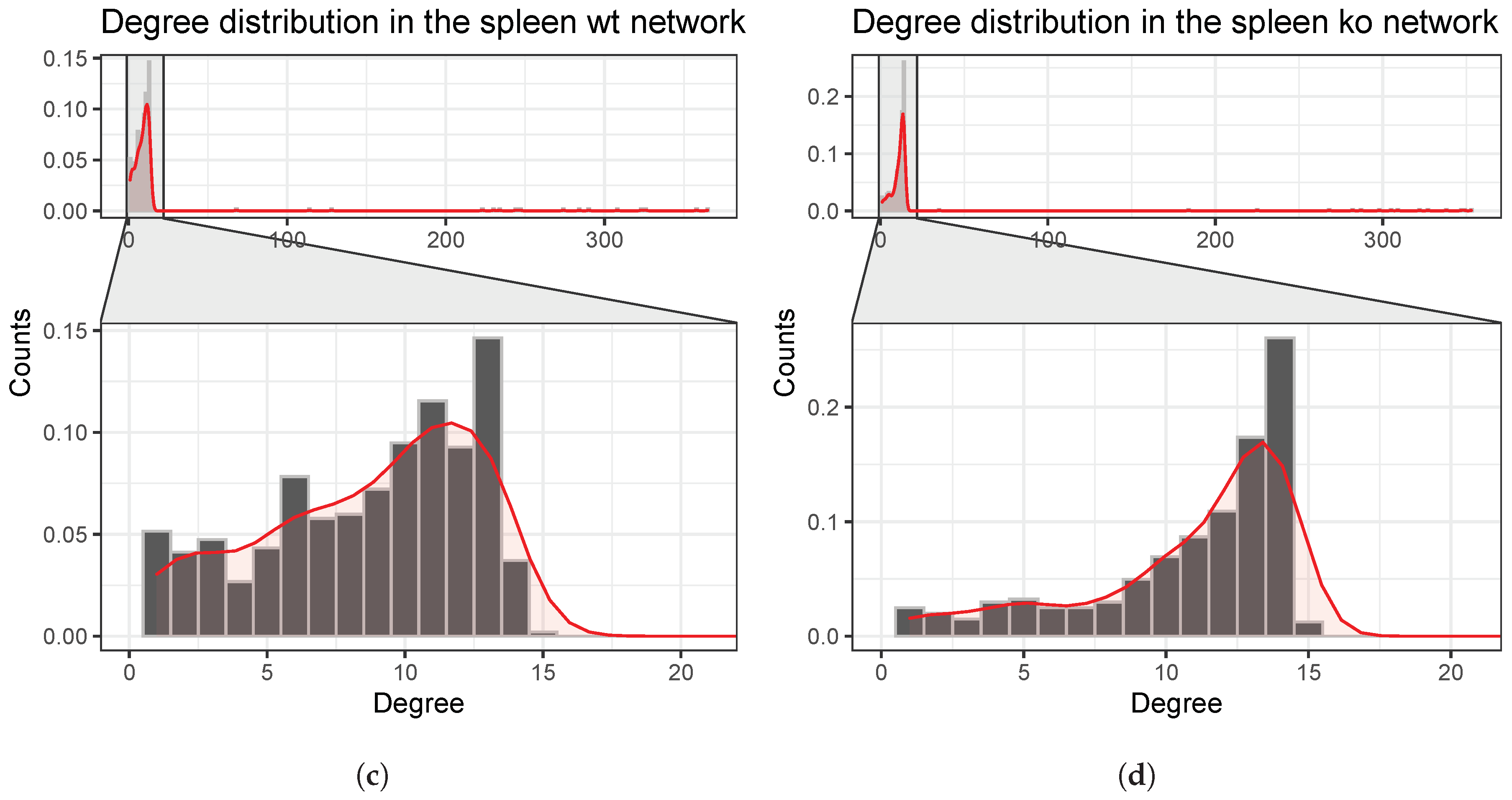
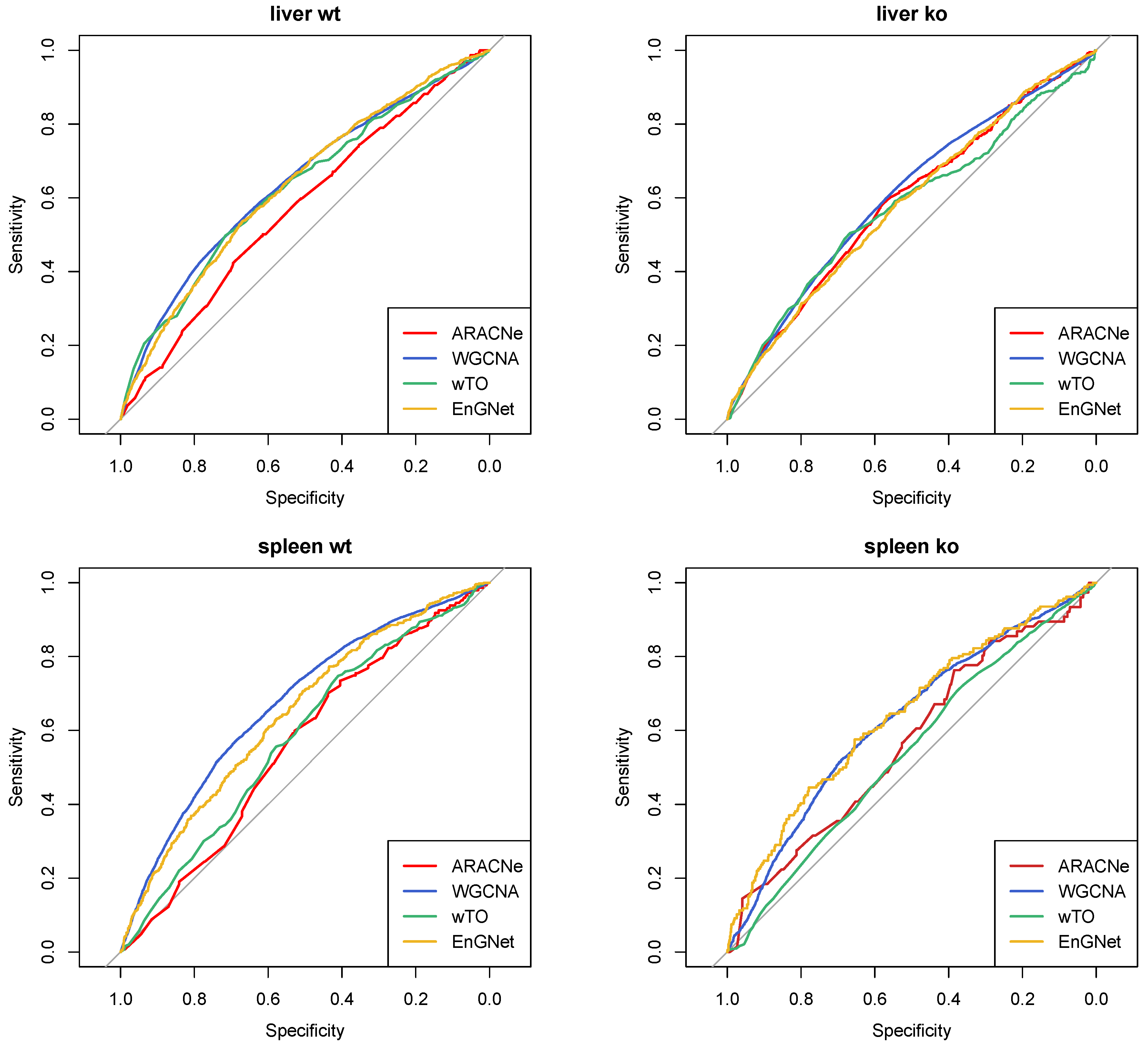
4.3. Reconstruction and Analysis of Gene Networks
5. Discussion
5.1. Exploratory Analyses Revealed a Time-Series Llike Behaviour on Raw Data, Assisting Network Reconstruction
5.2. Differential Expression Analyses Revealed Significant Changes between Wild Type and Knockout Samples
5.3. The Ablation of Ly6E in HSC Results in Impaired Immune Response as Predicted by Enrichment Analyses
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. Figures and Tables





{kind=link}
{kind=link}
{kind=link}
{kind=link}
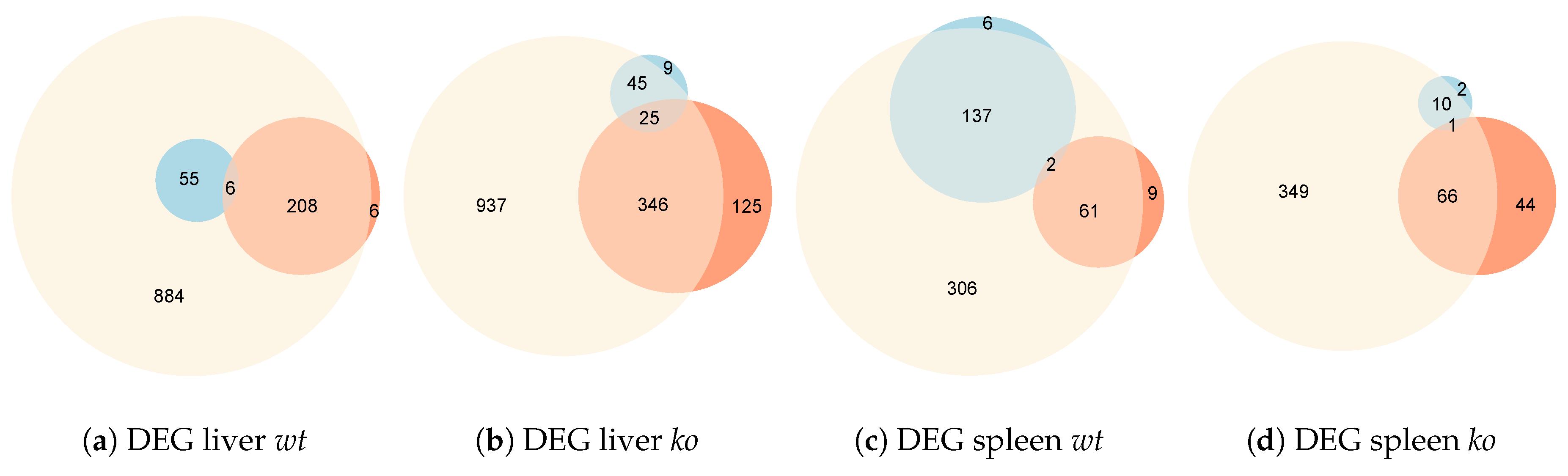{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liver wt | Liver ko | Spleen wt | Spleen ko | |
|---|---|---|---|---|
| Input genes | 1133 | 1153 | 506 | 426 |
| Network genes | 1118 | 1300 | 485 | 403 |
| Cluster 1 | 262 | 284 | 180 | 109 |
| Cluster 2 | 218 | 379 | 255 | 190 |
| Cluster 3 | 579 | 624 | 36 | 77 |
| Cluster 4 | 59 | 25 | ||
| Unconnected/minor clustered | 0 | 13 | 14 | 2 |






| Ensembl ID | Cluster | Degree | Reg. | Symbol | Description |
|---|---|---|---|---|---|
| ENSMUSG00000034593 | 1 | 1033 | up | Myo5a | myosin VA |
| ENSMUSG00000000982 | 3 | 1006 | up | Ccl3 | chemokine (C-C motif) ligand 3 |
| ENSMUSG00000030745 | 2 | 997 | up | Il21r | interleukin 21 receptor |
| ENSMUSG00000032322 | 3 | 989 | up | Pstpip1 | proline-serine-threonine phosphatase-interacting protein 1 |
| ENSMUSG00000079227 | 3 | 975 | up | Ccr5 | chemokine (C-C motif) receptor 5 |
| ENSMUSG00000031304 | 3 | 957 | up | Il2rg | interleukin 2 receptor, gamma chain |
| ENSMUSG00000069268 | 3 | 940 | up | Hist1h2bf | histone cluster 1, H2bf |
| ENSMUSG00000027071 | 1 | 938 | down | P2rx3 | purinergic receptor P2X, ligand-gated ion channel, 3 |
| ENSMUSG00000019232 | 3 | 929 | down | Etnppl | ethanolamine phosphate phospholyase |
| ENSMUSG00000032643 | 3 | 921 | up | Fhl3 | four and a half LIM domains 3 |
| ENSMUSG00000033763 | 3 | 904 | down | Mtss2 | MTSS I-BAR domain containing 2 |
| ENSMUSG00000032094 | 1 | 887 | up | Cd3d | CD3 antigen, delta polypeptide |
| ENSMUSG00000050896 | 3 | 883 | up | Rtn4rl2 | reticulon 4 receptor-like 2 |
| ENSMUSG00000067219 | 4 | 801 | down | Nipal1 | NIPA-like domain containing 1 |
| ENSMUSG00000110439 | 3 | 780 | down | Mup22 | major urinary protein 22 |
| ENSMUSG00000004105 | 2 | 743 | down | Angptl2 | angiopoietin-like 2 |
| ENSMUSG00000081650 | 1 | 713 | up | Gm16181 | - |
| ENSMUSG00000050395 | 2 | 538 | up | Tnfsf15 | tumor necrosis factor (ligand) superfamily, member 15 |
| ENSMUSG00000038067 | 1 | 220 | up | Csf3 | colony stimulating factor 3 (granulocyte) |
| ENSMUSG00000026104 | 2 | 90 | up | Stat1 | signal transducer and activator of transcription 1 |
| ENSMUSG00000037965 | 2 | 66 | up | Zc3h7a | zinc finger CCCH type containing 7 A |
| Ensembl ID | Cluster | Degree | Reg. | Symbol | Description |
|---|---|---|---|---|---|
| ENSMUSG00000029445 | 2 | 800 | down | Hpd | 4-hydroxyphenylpyruvic acid dioxygenase |
| ENSMUSG00000037071 | 3 | 781 | down | Scd1 | stearoyl-Coenzyme A desaturase 1 |
| ENSMUSG00000041773 | 3 | 773 | up | Enc1 | ectodermal-neural cortex 1 |
| ENSMUSG00000075015 | 3 | 760 | up | Gm10801 | - |
| ENSMUSG00000021250 | 3 | 742 | up | Fos | FBJ osteosarcoma oncogene |
| ENSMUSG00000031618 | 3 | 735 | down | Nr3c2 | nuclear receptor subfamily 3, group C, member 2 |
| ENSMUSG00000022419 | 1 | 732 | down | Deptor | DEP domain containing MTOR-interacting protein |
| ENSMUSG00000033610 | 3 | 700 | down | Pank1 | pantothenate kinase 1 |
| ENSMUSG00000024349 | 3 | 667 | up | Tmem173 | transmembrane protein 173 |
| ENSMUSG00000006519 | 3 | 666 | up | Cyba | cytochrome b-245, alpha polypeptide |
| ENSMUSG00000035878 | 3 | 666 | down | Hykk | hydroxylysine kinase 1 |
| ENSMUSG00000054630 | 2 | 652 | down | Ugt2b5 | UDP glucuronosyltransferase 2 family, polypeptide B5 |
| ENSMUSG00000041757 | 3 | 639 | down | Plekha6 | pleckstrin homology domain containing, family A member 6 |
| ENSMUSG00000053398 | 3 | 620 | up | Phgdh | 3-phosphoglycerate dehydrogenase |
| ENSMUSG00000022025 | 3 | 555 | down | Cnmd | chondromodulin |
| ENSMUSG00000029659 | 2 | 482 | up | Medag | mesenteric estrogen dependent adipogenesis |
| ENSMUSG00000062380 | 2 | 461 | up | Tubb3 | tubulin, beta 3 class III |
| ENSMUSG00000069309 | 3 | 408 | up | Hist1h2an | histone cluster 1, H2an |
| ENSMUSG00000034285 | 3 | 399 | down | Nipsnap1 | nipsnap homolog 1 |
| ENSMUSG00000027654 | 3 | 355 | up | Fam83d | family with sequence similarity 83, member D |
| ENSMUSG00000073435 | 2 | 355 | down | Nme3 | NME/NM23 nucleoside diphosphate kinase 3 |
| ENSMUSG00000021062 | 2 | 336 | up | Rab15 | RAB15, member RAS oncogene family |
| ENSMUSG00000037852 | 3 | 271 | up | Cpe | carboxypeptidase E |
| ENSMUSG00000096201 | 2 | 260 | up | Gm10715 | - |
| ENSMUSG00000022754 | 2 | 245 | up | Tmem45a | transmembrane protein 45a |
| ENSMUSG00000038233 | 1 | 239 | down | Gask1a | golgi associated kinase 1A |
| ENSMUSG00000043456 | 2 | 236 | up | Zfp536 | zinc finger protein 536 |
| ENSMUSG00000095891 | 2 | 168 | up | Gm10717 | - |
| ENSMUSG00000096688 | 1 | 126 | down | Mup17 | major urinary protein 17 |
| ENSMUSG00000099398 | 2 | 115 | up | Ms4a14 | membrane-spanning 4-domains, subfamily A, member 14 |
| ENSMUSG00000025002 | 1 | 99 | down | Cyp2c55 | cytochrome P450, family 2, subfamily c, polypeptide 55 |
| ENSMUSG00000074896 | 1 | 91 | up | Ifit3 | interferon-induced protein with tetratricopeptide repeats 3 |
| ENSMUSG00000062488 | 1 | 86 | up | Ifit3b | interferon-induced protein with tetratricopeptide repeats 3B |
| ENSMUSG00000029417 | 1 | 78 | up | Cxcl9 | chemokine (C-X-C motif) ligand 9 |
| ENSMUSG00000057465 | 1 | 77 | up | Saa2 | serum amyloid A 2 |
| ENSMUSG00000050908 | 2 | 69 | up | Tvp23a | trans-golgi network vesicle protein 23A |
| ENSMUSG00000030142 | 1 | 63 | up | Clec4e | C-type lectin domain family 4, member e |
| ENSMUSG00000038751 | 1 | 61 | down | Ptk6 | PTK6 protein tyrosine kinase 6 |
| ENSMUSG00000068606 | 1 | 40 | up | Gm4841 | predicted gene 4841 |
| Ensembl ID | Cluster | Degree | Reg. | Symbol | Description |
|---|---|---|---|---|---|
| ENSMUSG00000019505 | 2 | 365 | up | Ubb | ubiquitin B |
| ENSMUSG00000094777 | 2 | 358 | up | Hist1h2ap | histone cluster 1, H2ap |
| ENSMUSG00000057729 | 3 | 326 | up | Prtn3 | proteinase 3 |
| ENSMUSG00000056071 | 1 | 323 | up | S100a9 | S100 calcium binding protein A9 (calgranulin B) |
| ENSMUSG00000025403 | 2 | 308 | up | Shmt2 | serine hydroxymethyltransferase 2 (mitochondrial) |
| ENSMUSG00000023132 | 2 | 290 | up | Gzma | granzyme A |
| ENSMUSG00000078920 | 2 | 284 | up | Ifi47 | interferon gamma inducible protein 47 |
| ENSMUSG00000037894 | 1 | 274 | up | H2afz | H2A histone family, member Z |
| ENSMUSG00000035472 | 2 | 247 | down | Slc25a21 | solute carrier family 25 (mitochondrial oxodicarboxylate carrier), member 21 |
| ENSMUSG00000009350 | 1 | 244 | up | Mpo | myeloperoxidase |
| ENSMUSG00000103254 | 1 | 234 | up | Ighv1-15 | - |
| ENSMUSG00000069274 | 1 | 230 | up | Hist1h4f | histone cluster 1, H4f |
| ENSMUSG00000028328 | 2 | 223 | down | Tmod1 | tropomodulin 1 |
| ENSMUSG00000094322 | 1 | 128 | up | Ighv9-4 | - |
| ENSMUSG00000094124 | 1 | 114 | up | Ighv1-74 | - |
| ENSMUSG00000094546 | 1 | 68 | up | Ighv1-26 | - |
| Ensembl ID | Cluster | Degree | Reg. | Symbol | Description |
|---|---|---|---|---|---|
| ENSMUSG00000027715 | 2 | 353 | up | Ccna2 | cyclin A2 |
| ENSMUSG00000024742 | 3 | 349 | up | Fen1 | flap structure specific endonuclease 1 |
| ENSMUSG00000024640 | 2 | 347 | up | Psat1 | phosphoserine aminotransferase 1 |
| ENSMUSG00000040026 | 2 | 338 | up | Saa3 | serum amyloid A 3 |
| ENSMUSG00000039713 | 2 | 327 | down | Plekhg5 | pleckstrin homology domain containing, family G (with RhoGef domain) member 5 |
| ENSMUSG00000075289 | 4 | 322 | down | Carns1 | carnosine synthase 1 |
| ENSMUSG00000067610 | 2 | 309 | down | Klri1 | killer cell lectin-like receptor family I member 1 |
| ENSMUSG00000031503 | 1 | 305 | up | Col4a2 | collagen, type IV, alpha 2 |
| ENSMUSG00000095700 | 3 | 298 | up | Ighv10-3 | - |
| ENSMUSG00000076613 | 3 | 287 | up | Ighg2b | - |
| ENSMUSG00000051079 | 2 | 282 | down | Rgs13 | regulator of G-protein signaling 13 |
| ENSMUSG00000036027 | 2 | 268 | down | 1810046K07Rik | RIKEN cDNA 1810046K07 gene |
| ENSMUSG00000027962 | 1 | 225 | up | Vcam1 | vascular cell adhesion molecule 1 |
| ENSMUSG00000049130 | 1 | 184 | up | C5ar1 | complement component 5a receptor 1 |
| ENSMUSG00000066861 | 1 | 35 | up | Oas1g | 2′-5′ oligoadenylate synthetase 1G |
| Ensembl ID | Symbol | Description | Memb. | Reg. Type |
|---|---|---|---|---|
| ENSMUSG00000032487 | Ptgs2 | prostaglandin-endoperoxide synthase 2 | liver wt | 1 |
| ENSMUSG00000029816 | Gpnmb | glycoprotein (transmembrane) nmb | liver wt | 1 |
| ENSMUSG00000035385 | Ccl2 | chemokine (C-C motif) ligand 2 | liver wt | 1 |
| ENSMUSG00000035373 | Ccl7 | chemokine (C-C motif) ligand 7 | liver wt | 1 |
| ENSMUSG00000015437 | Gzmb | granzyme B | liver wt | 1 |
| ENSMUSG00000038037 | Socs1 | suppressor of cytokine signaling 1 | liver wt | 1 |
| ENSMUSG00000026839 | Upp2 | uridine phosphorylase 2 | liver ko | 2 |
| ENSMUSG00000075014 | Gm10800 | - | liver ko | 1 |
| ENSMUSG00000040660 | Cyp2b9 | cytochrome P450, family 2, subfamily b, polypeptide 9 | liver ko | 2 |
| ENSMUSG00000056978 | Hamp2 | hepcidin antimicrobial peptide 2 | liver ko | 2 |
| ENSMUSG00000073940 | Hbb-bt | hemoglobin, beta adult t chain | liver ko | 2 |
| ENSMUSG00000052305 | Hbb-bs | hemoglobin, beta adult major chain | liver ko | 2 |
| ENSMUSG00000025473 | Adam8 | a disintegrin and metallopeptidase domain 8 | liver ko | 1 |
| ENSMUSG00000056973 | Ces1d | carboxylesterase 1D | liver ko | 2 |
| ENSMUSG00000025317 | Car5a | carbonic anhydrase 5a, mitochondrial | liver ko | 2 |
| ENSMUSG00000050578 | Mmp13 | matrix metallopeptidase 13 | liver ko | 1 |
| ENSMUSG00000049723 | Mmp12 | matrix metallopeptidase 12 | liver ko | 1 |
| ENSMUSG00000035878 | Hykk | hydroxylysine kinase 1 | liver ko | 2 |
| ENSMUSG00000069917 | Hba-a2 | hemoglobin alpha, adult chain 2 | liver ko | 2 |
| ENSMUSG00000009350 | Mpo | myeloperoxidase | liver ko | 1 |
| ENSMUSG00000109482 | Gm4756 | - | liver ko | 2 |
| ENSMUSG00000060807 | Serpina6 | serine (or cysteine) peptidase inhibitor, clade A, member 6 | liver ko | 2 |
| ENSMUSG00000079018 | Ly6c1 | lymphocyte antigen 6 complex, locus C1 | liver ko | 1 |
| ENSMUSG00000074896 | Ifit3 | interferon-induced protein with tetratricopeptide repeats 3 | liver ko | 3 |
| ENSMUSG00000062488 | Ifit3b | interferon-induced protein with tetratricopeptide repeats 3B | liver ko | 3 |
| ENSMUSG00000032808 | Cyp2c38 | cytochrome P450, family 2, subfamily c, polypeptide 38 | liver ko | 2 |
| ENSMUSG00000025004 | Cyp2c40 | cytochrome P450, family 2, subfamily c, polypeptide 40 | liver ko | 2 |
| ENSMUSG00000042248 | Cyp2c37 | cytochrome P450, family 2, subfamily c, polypeptide 37 | liver ko | 2 |
| ENSMUSG00000067225 | Cyp2c54 | cytochrome P450, family 2, subfamily c, polypeptide 54 | liver ko | 2 |
| ENSMUSG00000054827 | Cyp2c50 | cytochrome P450, family 2, subfamily c, polypeptide 50 | liver ko | 2 |
| ENSMUSG00000001131 | Timp1 | tissue inhibitor of metalloproteinase 1 | liver ko | 1 |
| ENSMUSG00000015437 | Gzmb | granzyme B | spleen wt | 1 |
| ENSMUSG00000022584 | Ly6c2 | lymphocyte antigen 6 complex, locus C2 | spleen wt | 1 |
| ENSMUSG00000040026 | Saa3 | serum amyloid A 3 | spleen ko | 1 |

Appendix B. Validation of the Reconstruction Method


References
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [PubMed]
- Prentice, E.; McAuliffe, J.; Lu, X.; Subbarao, K.; Denison, M.R. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J. Virol. 2004, 78, 9977–9986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [Green Version]
- Voit, E. A First Course in Systems Biology; Garland Science: New York, NY, USA, 2017. [Google Scholar]
- Delgado, F.M.; Gómez-Vela, F. Computational methods for Gene Regulatory Networks reconstruction and analysis: A review. Artif. Intell. Med. 2019, 95, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vela, F.; Lagares, J.A.; Díaz-Díaz, N. Gene network coherence based on prior knowledge using direct and indirect relationships. Comput. Biol. Chem. 2015, 56, 142–151. [Google Scholar] [CrossRef]
- Hecker, M.; Lambeck, S.; Toepfer, S.; Van Someren, E.; Guthke, R. Gene regulatory network inference: Data integration in dynamic models—A review. Biosystems 2009, 96, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vela, F.; Rodriguez-Baena, D.S.; Vázquez-Noguera, J.L. Structure Optimization for Large Gene Networks Based on Greedy Strategy. Comput. Math. Methods Med. 2018, 2018, 9674108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Ding, Z.; Wan, L.; Tong, W.; Mao, J.; Li, L.; Hu, J.; Yang, M.; Liu, B.; Qian, X. Comprehensive analysis of the long noncoding RNA expression profile and construction of the lncRNA-mRNA co-expression network in colorectal cancer. Cancer Biol. Ther. 2020, 21, 157–169. [Google Scholar] [CrossRef]
- Díaz-Montaña, J.J.; Gómez-Vela, F.; Díaz-Díaz, N. GNC–app: A new Cytoscape app to rate gene networks biological coherence using gene–gene indirect relationships. Biosystems 2018, 166, 61–65. [Google Scholar] [CrossRef]
- Kumari, S.; Nie, J.; Chen, H.S.; Ma, H.; Stewart, R.; Li, X.; Lu, M.Z.; Taylor, W.M.; Wei, H. Evaluation of gene association methods for coexpression network construction and biological knowledge discovery. PLoS ONE 2012, 7, e50411. [Google Scholar] [CrossRef]
- de Siqueira Santos, S.; Takahashi, D.Y.; Nakata, A.; Fujita, A. A comparative study of statistical methods used to identify dependencies between gene expression signals. Brief. Bioinform. 2013, 15, 906–918. [Google Scholar] [CrossRef] [Green Version]
- Liesecke, F.; Daudu, D.; Dugé de Bernonville, R.; Besseau, S.; Clastre, M.; Courdavault, V.; de Craene, J.O.; Crèche, J.; Giglioli-Guivarc’h, N.; Glévarec, G.; et al. Ranking genome-wide correlation measurements improves microarray and RNA-seq based global and targeted co-expression networks. Sci. Rep. 2018, 8, 10885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Aderhold, A.; Bonneau, R.; Chen, Y.; et al. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Langfelder, P.; Horvath, S. Comparison of co-expression measures: Mutual information, correlation, and model based indices. BMC Bioinform. 2012, 13, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villaverde, A.F.; Ross, J.; Morán, F.; Banga, J.R. MIDER: Network inference with mutual information distance and entropy reduction. PLoS ONE 2014, 9, e96732. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Bai, J.; Yuan, C.; Long, L.; Zheng, Z.; Wang, Q.; Chenand, F.; Zhou, Y. Bioinformatics analysis and identification of potential genes related to pathogenesis of cervical intraepithelial neoplasia. J. Cancer 2020, 11, 2150–2157. [Google Scholar] [CrossRef]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A.; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2018, 115, E4179–E4188. [Google Scholar] [CrossRef] [Green Version]
- Sandor, C.; Beer, N.L.; Webber, C. Diverse type 2 diabetes genetic risk factors functionally converge in a phenotype-focused gene network. PLoS Comput. Biol. 2017, 13, e1005816. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Huang, J.; Jiang, M.; Sun, L. Survivin (BIRC5) cell cycle computational network in human no-tumor hepatitis/cirrhosis and hepatocellular carcinoma transformation. J. Cell. Biochem. 2011, 112, 1286–1294. [Google Scholar] [CrossRef]
- He, D.; Liu, Z.P.; Honda, M.; Kaneko, S.; Chen, L. Coexpression network analysis in chronic hepatitis B and C hepatic lesions reveals distinct patterns of disease progression to hepatocellular carcinoma. J. Mol. Cell Biol. 2012, 4, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Nogales, A.; Martínez-Sobrido, L. Reverse genetics approaches for the development of influenza vaccines. Int. J. Mol. Sci. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.H.; Fung, T.S.; Fang, S.; Huang, M.; Le, M.T.; Liu, D.X. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology 2018, 515, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; McGarvey, P.; Madhavan, S.; Kumar, R.; Gusev, Y.; Upadhyay, G. Distinct lymphocyte antigens 6 (Ly6) family members Ly6D, Ly6E, Ly6K and Ly6H drive tumorigenesis and clinical outcome. Oncotarget 2016, 7, 11165. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liu, S.L. Emerging Role of LY6E in Virus–Host Interactions. Viruses 2019, 11, 1020. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.C.; Niikura, M.; Fulton, J.; Cheng, H. Identification of chicken lymphocyte antigen 6 complex, locus E (LY6E, alias SCA2) as a putative Marek’s disease resistance gene via a virus-host protein interaction screen. Cytogenet. Genome Res. 2003, 102, 304–308. [Google Scholar] [CrossRef]
- Stier, M.T.; Spindler, K.R. Polymorphisms in Ly6 genes in Msq1 encoding susceptibility to mouse adenovirus type 1. Mamm. Genome 2012, 23, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Liang, C.; Liu, S.L. Interferon-inducible LY6E protein promotes HIV-1 infection. J. Biol. Chem. 2017, 292, 4674–4685. [Google Scholar] [CrossRef] [Green Version]
- Mar, K.B.; Rinkenberger, N.R.; Boys, I.N.; Eitson, J.L.; McDougal, M.B.; Richardson, R.B.; Schoggins, J.W. LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Hackett, B.A.; Cherry, S. Flavivirus internalization is regulated by a size-dependent endocytic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 4246–4251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Vela, F.; Delgado-Chaves, F.M.; Rodríguez-Baena, D.S.; García-Torres, M.; Divina, F. Ensemble and Greedy Approach for the Reconstruction of Large Gene Co-Expression Networks. Entropy 2019, 21, 1139. [Google Scholar] [CrossRef] [Green Version]
- Giulietti, M.; Occhipinti, G.; Principato, G.; Piva, F. Identification of candidate miRNA biomarkers for pancreatic ductal adenocarcinoma by weighted gene co-expression network analysis. Cell. Oncol. 2017, 40, 181–192. [Google Scholar] [CrossRef]
- Ray, S.; Hossain, S.M.M.; Khatun, L.; Mukhopadhyay, A. A comprehensive analysis on preservation patterns of gene co-expression networks during Alzheimer’s disease progression. BMC Bioinform. 2017, 18, 579. [Google Scholar] [CrossRef]
- Medina, I.R.; Lubovac-Pilav, Z. Gene co-expression network analysis for identifying modules and functionally enriched pathways in type 1 diabetes. PLoS ONE 2016, 11, e0156006. [Google Scholar]
- van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Argilaguet Marqués, J.; Pedragosa Marín, M.; Esteve-Codina, A.; Riera Domínguez, M.G.; Vidal, E.; Peligero Cruz, C.; Casella, V.; Andreu Martínez, D.; Kaisho, T.; Bocharov, G.A.; et al. Systems analysis reveals complex biological processes during virus infection fate decisions. Genome Res. 2019, 29, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobadi, M.Z.; Mozhgani, S.H.; Farzanehpour, M.; Behzadian, F. Identifying novel biomarkers of the pediatric influenza infection by weighted co-expression network analysis. Virol. J. 2019, 16, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michlmayr, D.; Pak, T.R.; Rahman, A.H.; Amir, E.A.D.; Kim, E.Y.; Kim-Schulze, S.; Suprun, M.; Stewart, M.G.; Thomas, G.P.; Balmaseda, A.; et al. Comprehensive innate immune profiling of chikungunya virus infection in pediatric cases. Mol. Syst. Biol. 2018, 14, e7862. [Google Scholar] [CrossRef]
- Pedragosa, M.; Riera, G.; Casella, V.; Esteve-Codina, A.; Steuerman, Y.; Seth, C.; Bocharov, G.; Heath, S.C.; Gat-Viks, I.; Argilaguet, J.; et al. Linking cell dynamics with gene coexpression networks to characterize key events in chronic virus infections. Front. Immunol. 2019, 10, 1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Hossain, S.M.M.; Khatun, L. Discovering preservation pattern from co-expression modules in progression of HIV-1 disease: An eigengene based approach. In Proceedings of the 2016 International Conference on Advances in Computing, Communications and Informatics (ICACCI), Jaipur, India, 21–24 September 2016; pp. 814–820. [Google Scholar]
- McDermott, J.; Mitchell, H.; Gralinski, L.; Eisfeld, A.J.; Josset, L.; Bankhead, A., 3rd; Neumann, G.; Tilton, S.C.; Schäfer, A.; Li, C.; et al. The effect of inhibition of PP1 and TNFα signaling on pathogenesis of SARS coronavirus. BMC Syst. Biol. 2016, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, K.; Wang, Y.; Pan, P.; Xu, G.; Mo, L.; Cao, L.; Wu, C.; Shen, X. The regulatory role of microRNA-mRNA co-expression in hepatitis B virus-associated acute liver failure. Ann. Hepatol. 2019, 18, 883–892. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Albuquerque, N.; Baig, E.; Ma, X.; Zhang, J.; He, W.; Rowe, A.; Habal, M.; Liu, M.; Shalev, I.; Downey, G.P.; et al. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 2006, 80, 10382–10394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Fang, L.; Yuan, S.; Zhao, L.; Wang, X.; Long, S.; Wang, M.; Wang, D.; Foda, M.F.; Xiao, S. The nucleocapsid proteins of mouse hepatitis virus and severe acute respiratory syndrome coronavirus share the same IFN-β antagonizing mechanism: Attenuation of PACT-mediated RIG-I/MDA5 activation. Oncotarget 2017, 8, 49655. [Google Scholar] [CrossRef]
- Case, J.B.; Li, Y.; Elliott, R.; Lu, X.; Graepel, K.W.; Sexton, N.R.; Smith, E.C.; Weiss, S.R.; Denison, M.R. Murine hepatitis virus nsp14 exoribonuclease activity is required for resistance to innate immunity. J. Virol. 2018, 92, e01531-17. [Google Scholar] [CrossRef] [Green Version]
- Gorman, M.J.; Poddar, S.; Farzan, M.; Diamond, M.S. The interferon-stimulated gene Ifitm3 restricts West Nile virus infection and pathogenesis. J. Virol. 2016, 90, 8212–8225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loughner, C.; Bruford, E.; McAndrews, M.; Delp, E.E.; Swamynathan, S.; Swamynathan, S.K. Organization, evolution and functions of the human and mouse Ly6/uPAR family genes. Hum. Genom. 2016, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mar, K.B.; Eitson, J.; Schoggins, J. Interferon-stimulated gene LY6E enhances entry of diverse RNA viruses. J. Immunol. 2016, 196, 217.7. [Google Scholar]
- Giotis, E.S.; Robey, R.C.; Skinner, N.G.; Tomlinson, C.D.; Goodbourn, S.; Skinner, M.A. Chicken interferome: Avian interferon-stimulated genes identified by microarray and RNA-seq of primary chick embryo fibroblasts treated with a chicken type I interferon (IFN-α). Vet. Res. 2016, 47, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Mishra, B.; Mehmood, A.; Athar, M.; Mukhtar, M.S. Integrative Network Biology Framework Elucidates Molecular Mechanisms of SARS-CoV-2 Pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pfaender, S.; Mar, K.B.; Michailidis, E.; Kratzel, A.; Hirt, D.; V’kovski, P.; Fan, W.; Ebert, N.; Stalder, H.; Kleine-Weber, H.; et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115. [Google Scholar] [CrossRef]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef]
- Alvarez, J.M.; Riveras, E.; Vidal, E.A.; Gras, D.E.; Contreras-López, O.; Tamayo, K.P.; Aceituno, F.; Gómez, I.; Ruffel, S.; Lejay, L.; et al. Systems approach identifies TGA 1 and TGA 4 transcription factors as important regulatory components of the nitrate response of A rabidopsis thaliana roots. Plant J. 2014, 80, 1–13. [Google Scholar] [CrossRef]
- Delgado-Chaves, F.M.; Gómez-Vela, F.; García-Torres, M.; Divina, F.; Vázquez Noguera, J.L. Computational Inference of Gene Co-Expression Networks for the identification of Lung Carcinoma Biomarkers: An Ensemble Approach. Genes 2019, 10, 962. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Lopez, O.; Moyano, T.C.; Soto, D.C.; Gutiérrez, R.A. Step-by-step construction of gene co-expression networks from high-throughput arabidopsis RNA sequencing data. In Root Development; Springer: Berlin/Heidelberg, Germany, 2018; pp. 275–301. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Genovese, C.R.; Roeder, K.; Wasserman, L. False discovery control with p-value weighting. Biometrika 2006, 93, 509–524. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsen, J.A.; Pai, S.; Isserlin, R.; Demchak, B.; Pico, A.R. RCy3: Network biology using Cytoscape from within R. F1000Research 2019, 8, 1774. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, M.; Sun, J.; Wang, Y.; Jiang, R. Gene co-opening network deciphers gene functional relationships. Mol. Biosyst. 2017, 13, 2428–2439. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Kuchinsky, A.; Morris, J.H.; States, D.J.; Meng, F. GLay: Community structure analysis of biological networks. Bioinformatics 2010, 26, 3135–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doncheva, N.T.; Assenov, Y.; Domingues, F.S.; Albrecht, M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012, 7, 670. [Google Scholar] [CrossRef] [PubMed]
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR–G-protein binding. Nature 2017, 545, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Dovoedo, Y.; Chakraborti, S. Boxplot-based outlier detection for the location-scale family. Commun. Stat. Simul. Comput. 2015, 44, 1492–1513. [Google Scholar] [CrossRef]
- Yang, J.; Rahardja, S.; Fränti, P. Outlier detection: How to threshold outlier scores? In Proceedings of the International Conference on Artificial Intelligence, Information Processing and Cloud Computing, Sanya, China, 19–21 December 2019; pp. 1–6. [Google Scholar]
- Consortium, G.O. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef]
- Gibson, S.M.; Ficklin, S.P.; Isaacson, S.; Luo, F.; Feltus, F.A.; Smith, M.C. Massive-scale gene co-expression network construction and robustness testing using random matrix theory. PLoS ONE 2013, 8, e55871. [Google Scholar] [CrossRef] [Green Version]
- Milenković, T.; Pržulj, N. Uncovering biological network function via graphlet degree signatures. Cancer Inform. 2008, 6, CIN-S680. [Google Scholar] [CrossRef]
- Baron, S.; Fons, M.; Albrecht, T. Viral pathogenesis. In Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Deng, X.; Chen, Y.; Mielech, A.M.; Hackbart, M.; Kesely, K.R.; Mettelman, R.C.; O’Brien, A.; Chapman, M.E.; Mesecar, A.D.; Baker, S.C. Structure-Guided Mutagenesis Alters Deubiquitinating Activity and Attenuates Pathogenesis of a Murine Coronavirus. J. Virol. 2020. [Google Scholar] [CrossRef]
- Khan, H.A.; Ahmad, M.Z.; Khan, J.A.; Arshad, M.I. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 245–256. [Google Scholar] [CrossRef]
- Wu, D.; Wang, H.; Yan, W.; Chen, T.; Wang, M.; Han, M.; Wu, Z.; Wang, X.; Ai, G.; Xi, D.; et al. A disparate subset of double-negative T cells contributes to the outcome of murine fulminant viral hepatitis via effector molecule fibrinogen-like protein 2. Immunol. Res. 2016, 64, 518–530. [Google Scholar] [CrossRef]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Kim, J.Y.; Kim, H.S.; Oh, J.C.; Cheon, Y.H.; Park, J.; Yoon, K.H.; Lee, M.S.; Youn, B.S. Progranulin and a five transmembrane domain-containing receptor-like gene are the key components in receptor activator of nuclear factor κB (RANK)-dependent formation of multinucleated osteoclasts. J. Biol. Chem. 2015, 290, 2042–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Frattini, P.; Villa, C.; De Santis, F.; Meregalli, M.; Belicchi, M.; Erratico, S.; Bella, P.; Raimondi, M.T.; Lu, Q.; Torrente, Y. Autologous intramuscular transplantation of engineered satellite cells induces exosome-mediated systemic expression of Fukutin-related protein and rescues disease phenotype in a murine model of limb-girdle muscular dystrophy type 2I. Hum. Mol. Genet. 2017, 26, 3682–3698. [Google Scholar] [CrossRef] [Green Version]
- Desmedt, S.; Desmedt, V.; Delanghe, J.; Speeckaert, R.; Speeckaert, M. The intriguing role of soluble urokinase receptor in inflammatory diseases. Crit. Rev. Clin. Lab. Sci. 2017, 54, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zheng, S.; Chen, D.; Zheng, M.; Li, X.; Li, G.; Lin, H.; Chang, J.; Zeng, H.; Guo, J.T. LY6E Restricts the Entry of Human Coronaviruses, including the currently pandemic SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- yWorks. Available online: https://www.yworks.com/ (accessed on 16 July 2020).
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. In BMC Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2006; Volume 7, p. S7. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Gysi, D.M.; Voigt, A.; de Miranda Fragoso, T.; Almaas, E.; Nowick, K. WTO: An R package for computing weighted topological overlap and a consensus network with integrated visualization tool. BMC Bioinform. 2018, 19, 392. [Google Scholar] [CrossRef]
- Meyer, P.E.; Lafitte, F.; Bontempi, G. minet: AR/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinform. 2008, 9, 461. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4. [Google Scholar] [CrossRef] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Chaves, F.M.; Gómez-Vela, F.; Divina, F.; García-Torres, M.; Rodriguez-Baena, D.S. Computational Analysis of the Global Effects of Ly6E in the Immune Response to Coronavirus Infection Using Gene Networks. Genes 2020, 11, 831. https://doi.org/10.3390/genes11070831
Delgado-Chaves FM, Gómez-Vela F, Divina F, García-Torres M, Rodriguez-Baena DS. Computational Analysis of the Global Effects of Ly6E in the Immune Response to Coronavirus Infection Using Gene Networks. Genes. 2020; 11(7):831. https://doi.org/10.3390/genes11070831
Chicago/Turabian StyleDelgado-Chaves, Fernando M., Francisco Gómez-Vela, Federico Divina, Miguel García-Torres, and Domingo S. Rodriguez-Baena. 2020. "Computational Analysis of the Global Effects of Ly6E in the Immune Response to Coronavirus Infection Using Gene Networks" Genes 11, no. 7: 831. https://doi.org/10.3390/genes11070831
APA StyleDelgado-Chaves, F. M., Gómez-Vela, F., Divina, F., García-Torres, M., & Rodriguez-Baena, D. S. (2020). Computational Analysis of the Global Effects of Ly6E in the Immune Response to Coronavirus Infection Using Gene Networks. Genes, 11(7), 831. https://doi.org/10.3390/genes11070831