Extensive Genetic Connectivity and Historical Persistence Are Features of Two Widespread Tree Species in the Ancient Pilbara Region of Western Australia

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Species
2.2. Sampling
2.3. Microsatellite Data
2.4. Microsatellite Analysis
2.5. Chloroplast DNA (cpDNA) Amplification and Sequencing
2.6. cpDNA Analysis
3. Results
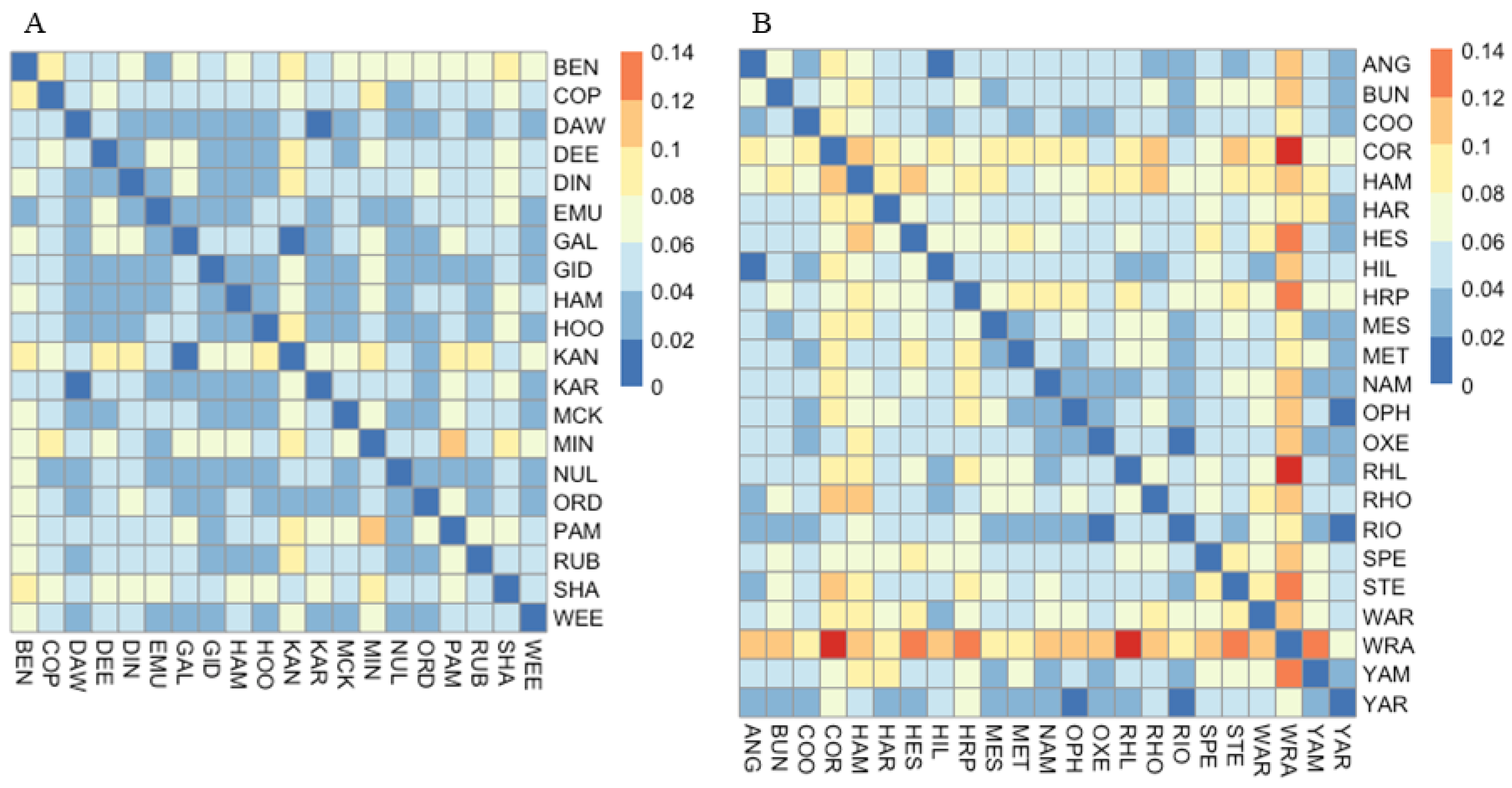
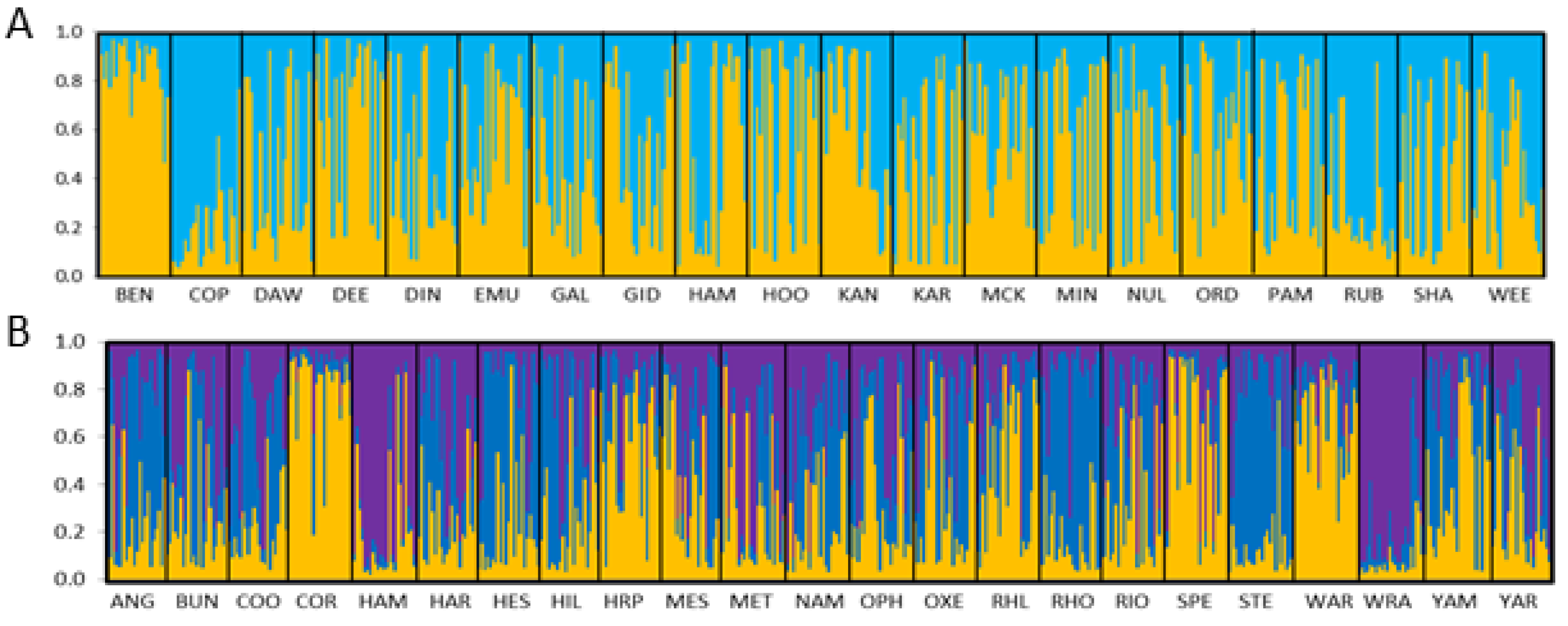
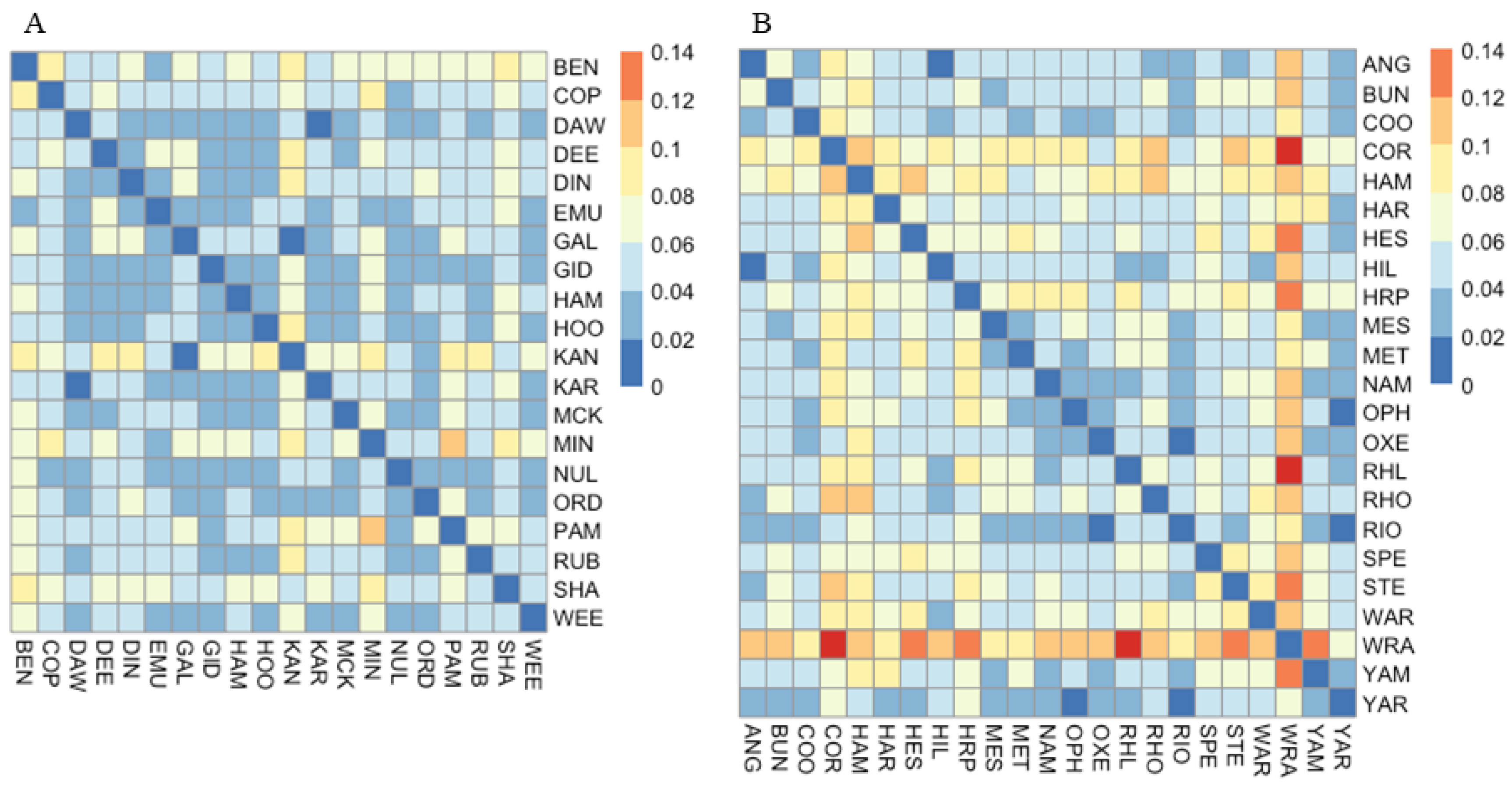
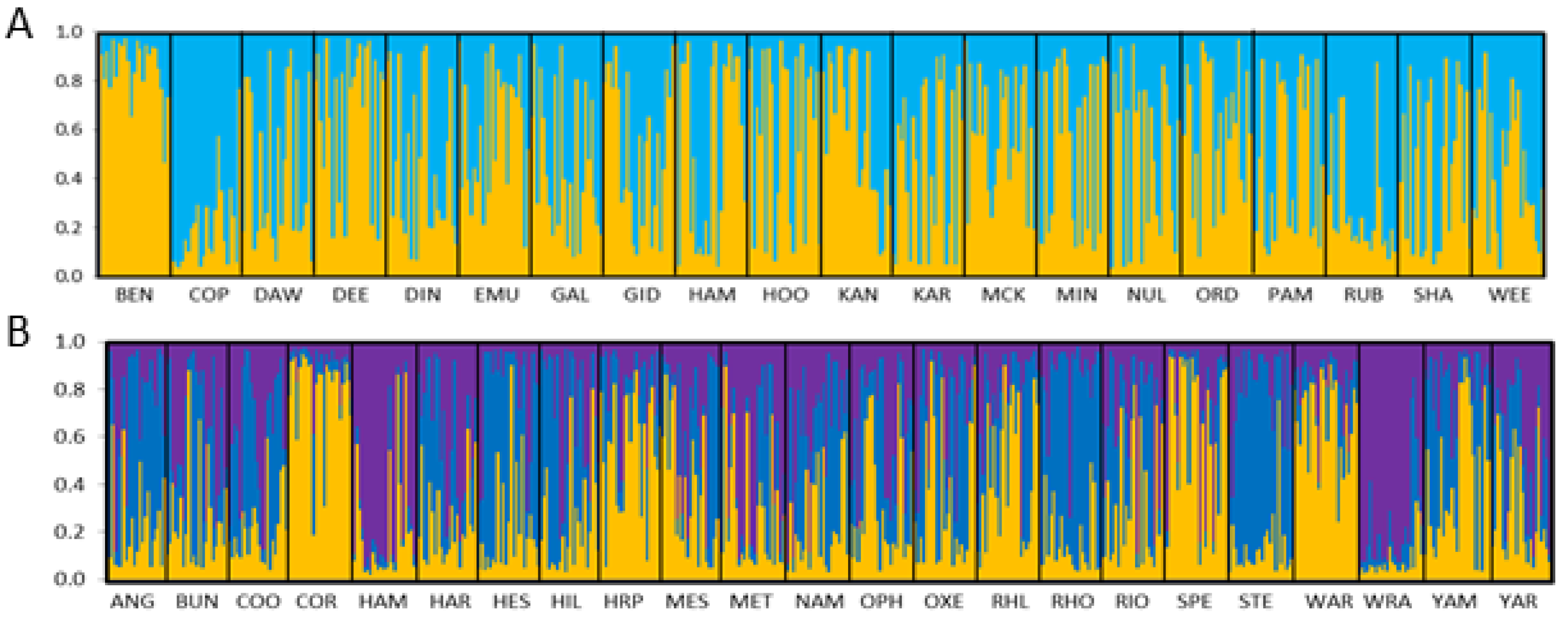
3.1. Microsatellite Data
3.1.1. Corymbia hamersleyana
3.1.2. Acacia pruinocarpa
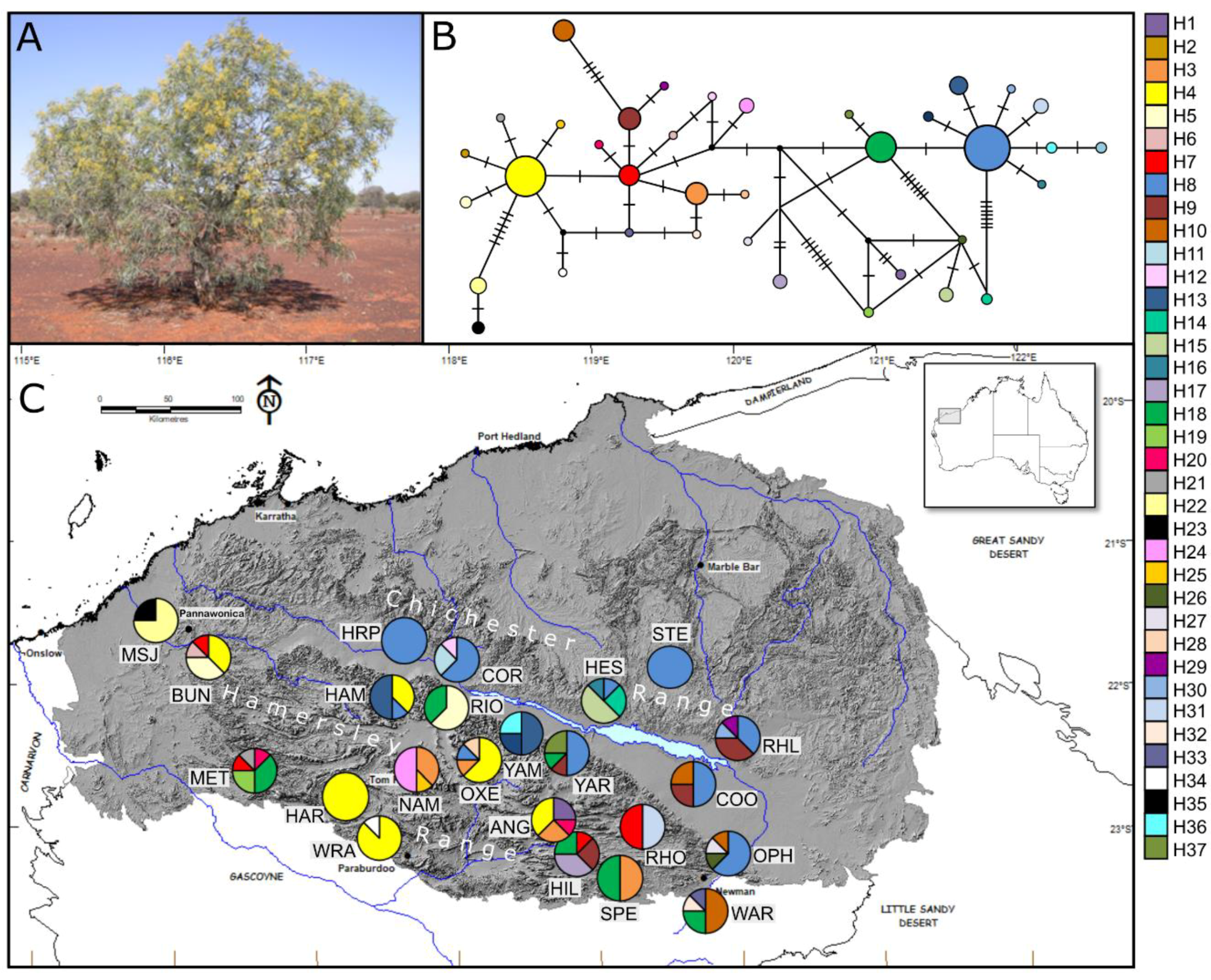
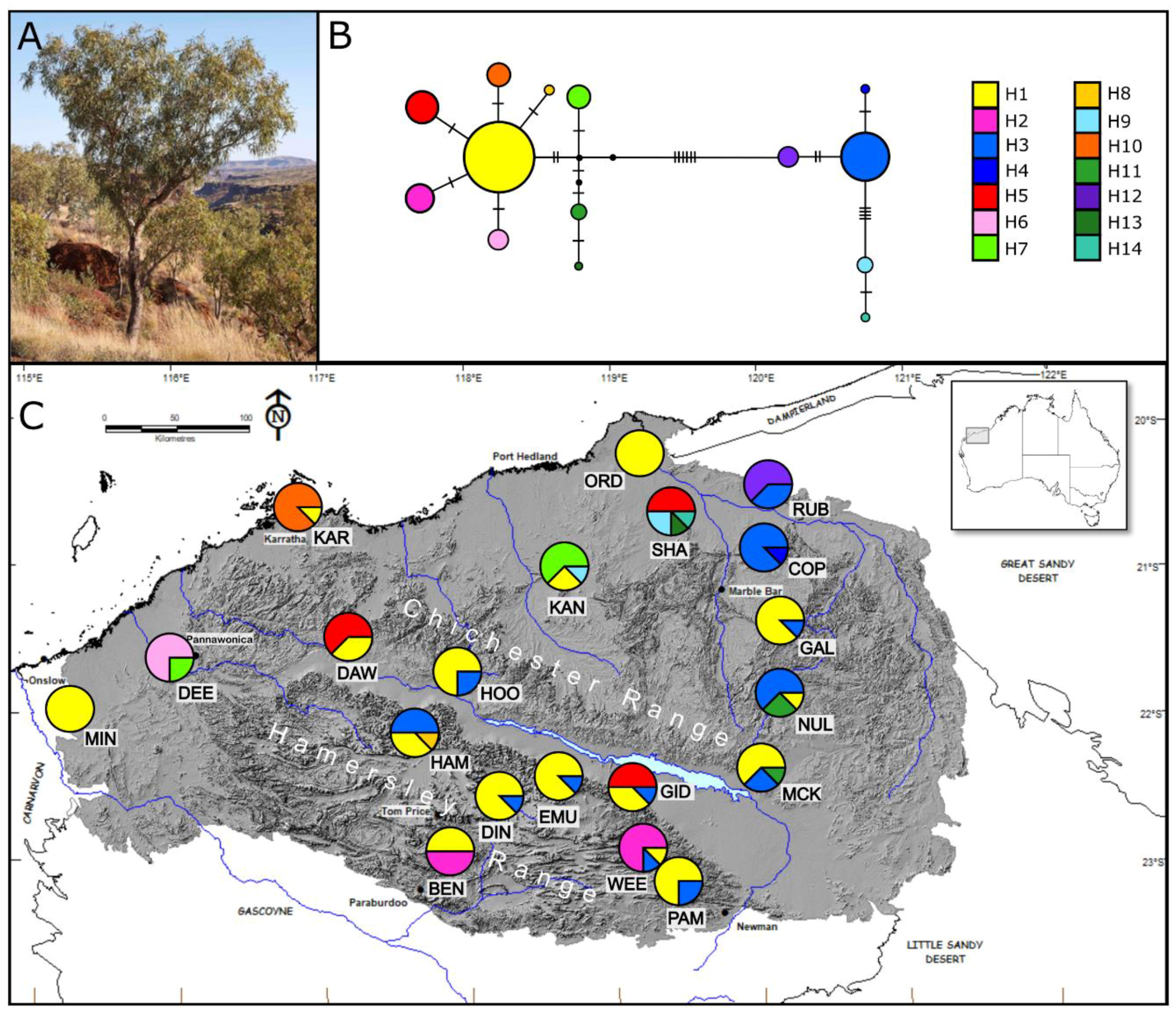
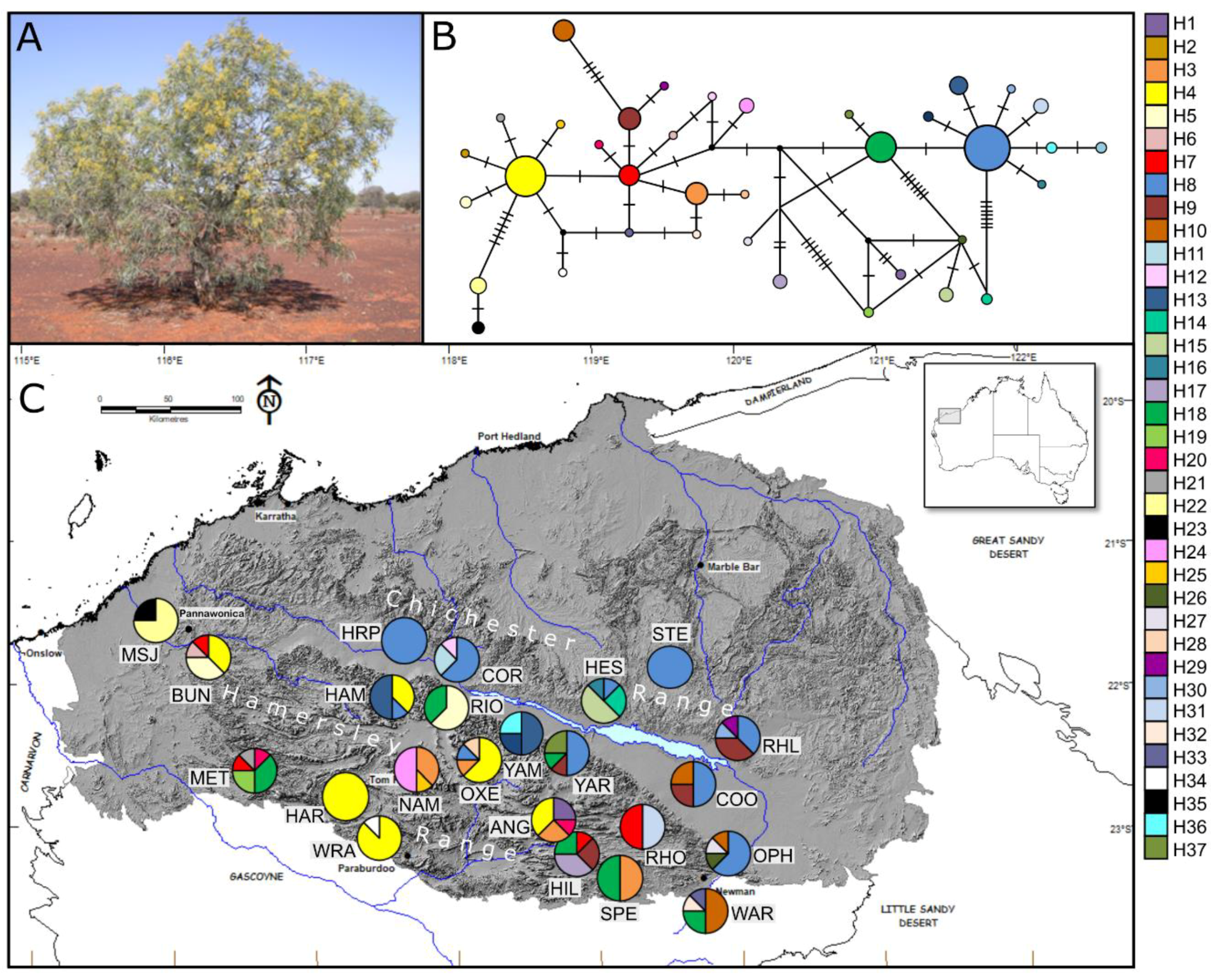
3.2. cpDNA Data
3.2.1. Corymbia hamersleyana
3.2.2. Acacia pruniocarpa
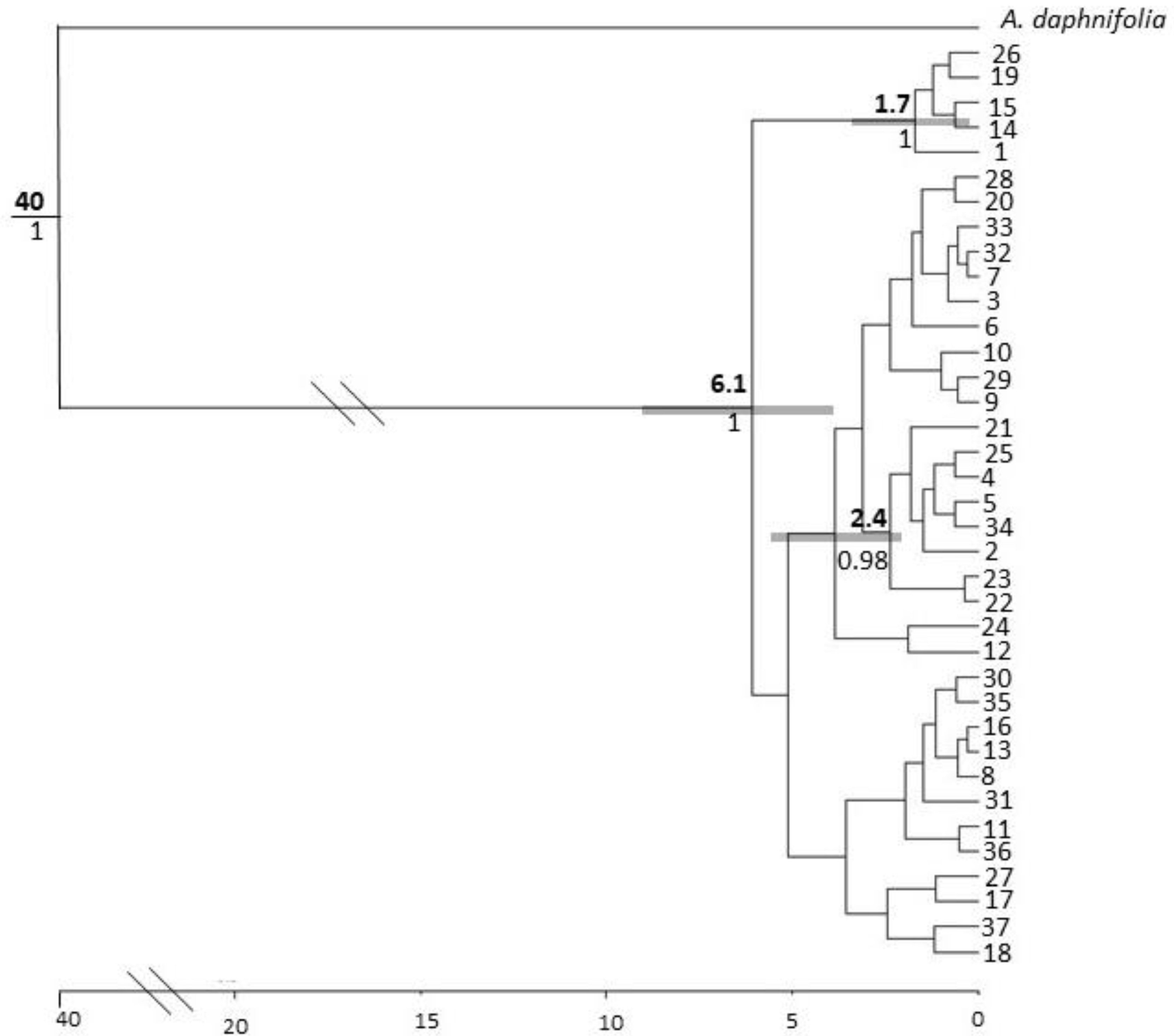
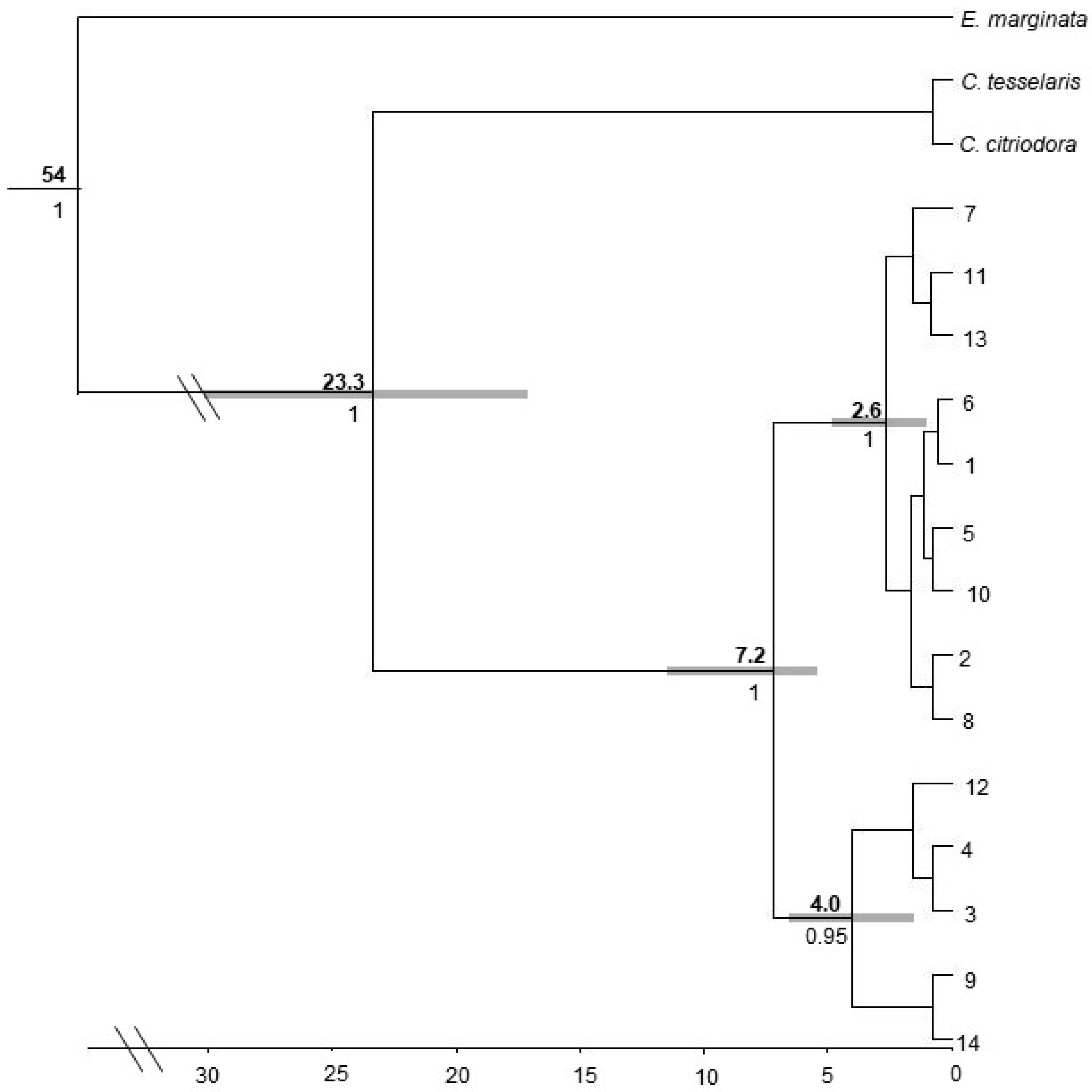
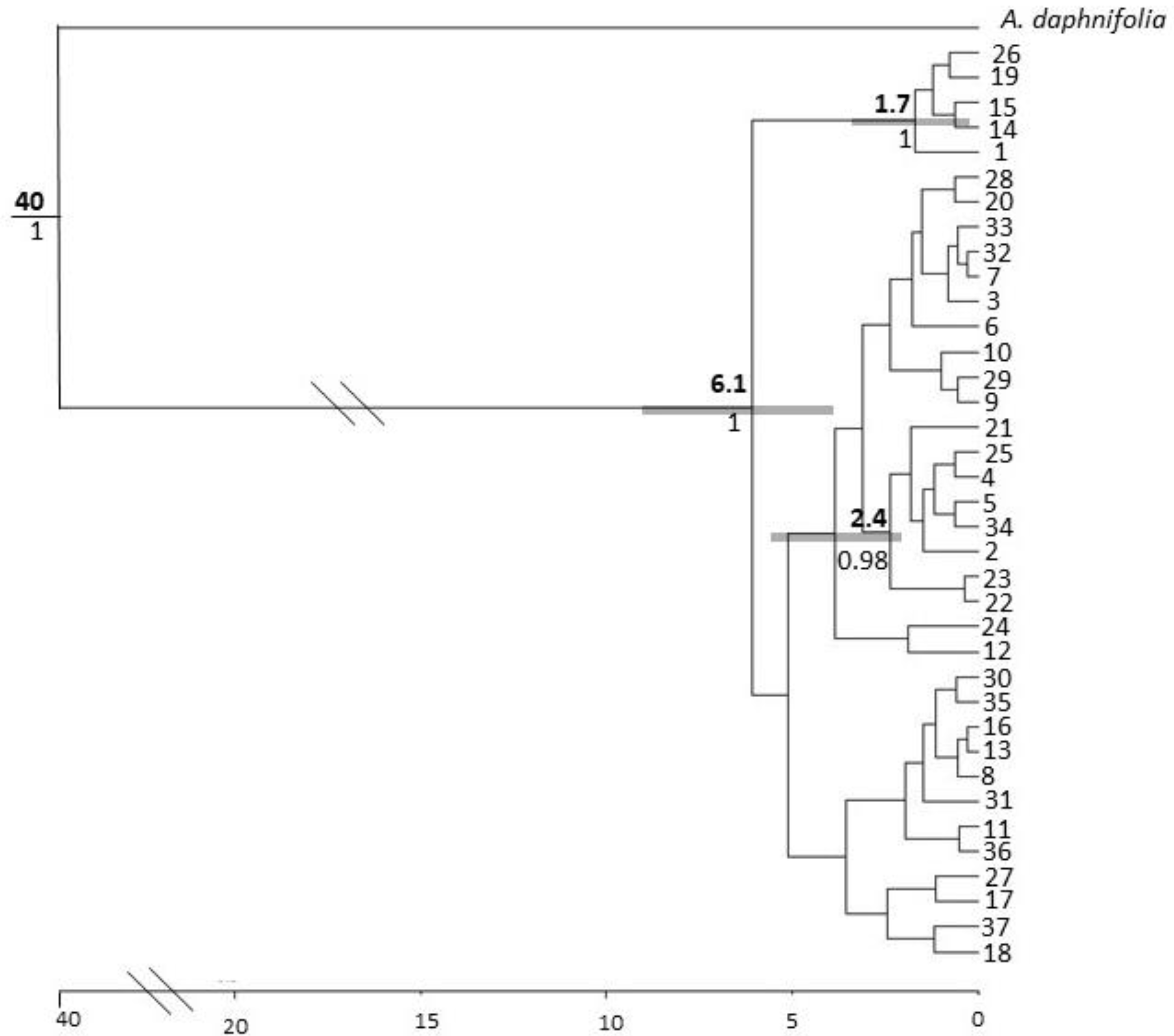
3.3. Molecular Dating and Evolutionary Relationships
3.3.1. Corymbia hamersleyana
3.3.2. Acacia pruinocarpa
4. Discussion
4.1. Historical Persistence
4.2. Topographic Refugia
4.3. Genetic Connectivity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hewitt, G.M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 1999, 68, 87–112. [Google Scholar] [CrossRef]
- Gomez, A.; Lunt, D.H. Refugia within refugia: Patterns of phylogeographic concordance in the Iberian Peninsula. In Phylogeography of Southern European Refugia; Weiss, S., Ferrand, N., Eds.; Springer: Amsterdam, The Netherlands, 2007; pp. 155–188. [Google Scholar]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.-G.; Cosson, J.-F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Potts, A.J.; Hedderson, T.A.; Cowling, R.M. Testing large-scale conservation corridors designed for patterns and processes: Comparative phylogeography of three tree species. Divers. Distrib. 2013, 19, 1418–1428. [Google Scholar] [CrossRef]
- Sork, V.L.; Gugger, P.F.; Chen, J.-M.; Werth, S. Evolutionary lessons from California plant phylogeography. Proc. Natl. Acad. Sci. USA 2016, 113, 8064–8071. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M. Phylogeography provides an evolutionary context for the conservation of a diverse and ancient flora. Aust. J. Bot. 2007, 55, 316–325. [Google Scholar] [CrossRef]
- Byrne, M. Evidence for multiple refugia at different time scales during Pleistocene climatic oscillations in southern Australia inferred from phylogeography. Quat. Sci. Rev. 2008, 27, 2576–2585. [Google Scholar] [CrossRef]
- Soltis, D.E.; Morris, A.B.; McLachlan, J.S.; Manos, P.S.; Soltis, P.S. Comparative phylogeography of unglaciated eastern North America. Mol. Ecol. 2006, 15, 4261–4293. [Google Scholar]
- Cowling, R.M.; Lombard, A.T. Heterogeneity, speciation/extinction history and climate: Explaining regional plant diversity patterns in the Cape Floristic Region. Divers. Distrib. 2002, 8, 163–179. [Google Scholar] [CrossRef]
- Harrison, S.; Noss, R. Endemism hotspots are linked to stable climatic refugia. Ann. Bot. 2017, 119, 207–214. [Google Scholar] [CrossRef]
- Nistelberger, H.M.; Gibson, N.; Macdonald, B.; Tapper, S.-L.; Byrne, M. Phylogeographic evidence for two mesic refugia in a biodiversity hotspot. Heredity (Edinb) 2014, 113, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M.; Yeates, D.K.; Joseph, L.; Kearney, M.; Bowler, J.; Williams, M.A.J.; Cooper, S.; Donnellan, S.C.; Keogh, J.S.; Leys, R.; et al. Birth of a biome: Insights into the assembly and maintenance of the Australian arid zone biota. Mol. Ecol. 2008, 17, 4398–4417. [Google Scholar] [CrossRef] [PubMed]
- Dalmaris, E.; Ramalho, C.E.; Poot, P.; Veneklaas, E.J.; Byrne, M. A climate change context for the decline of a foundation tree species in south-western Australia: Insights from phylogeography and species distribution modelling. Ann. Bot. 2015, 116, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, D.; Tapper, S.L.; Coates, D.; Mcarthur, S.; Hankinson, M.; Byrne, M. The role of fire and a long-lived soil seed bank in maintaining persistence, genetic diversity and connectivity in a fire-prone landscape. J. Biogeogr. 2016, 43, 70–84. [Google Scholar] [CrossRef]
- Byrne, M.; Coates, D.J.; Forest, F.; Hopper, S.D.; Krauss, S.L.; Sniderman, J.M.K.; Thiele, K.R. A diverse flora—Species and genetic relationships. In Plant Life on the Sandplains in Southwest Australia; Lambers, H., Ed.; UWA Publishing: Perth, Australia, 2014; pp. 81–99. [Google Scholar]
- Sampson, J.; Tapper, S.; Coates, D.; Hankinson, M.; McArthur, S.; Byrne, M. Persistence with episodic range expansion from the early Pleistocene: The distribution of genetic variation in the forest tree Corymbia calophylla (Myrtaceae) in south-western Australia. Biol. J. Linn. Soc. 2018, 123, 545–560. [Google Scholar] [CrossRef]
- Byrne, M.; Macdonald, B.; Brand, J. Phylogeography and divergence in the chloroplast genome of Western Australian Sandalwood (Santalum spicatum). Heredity (Edinb) 2003, 91, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, D.; Tapper, S.L.; Coates, D.; Hankinson, M.; Mcarthur, S.; Byrne, M. How does the post-fire facultative seeding strategy impact genetic variation and phylogeographical history? The case of Bossiaea ornata (Fabaceae) in a fire-prone, mediterranean-climate ecosystem. J. Biogeogr. 2016, 43, 96–110. [Google Scholar] [CrossRef]
- Llorens, T.M.; Tapper, S.L.; Coates, D.J.; McArthur, S.; Hankinson, M.; Byrne, M. Does population distribution matter? Influence of a patchy versus continuous distribution on genetic patterns in a wind-pollinated shrub. J. Biogeogr. 2017, 44, 361–374. [Google Scholar] [CrossRef]
- Levy, E.; Byrne, M.; Coates, D.J.; Macdonald, B.M.; McArthur, S.; Van Leeuwen, S. Contrasting influences of geographic range and distribution of populations on patterns of genetic diversity in two sympatric Pilbara acacias. PLoS ONE 2016, 11, 1–18. [Google Scholar] [CrossRef]
- Byrne, M.; Millar, M.A.; Coates, D.J.; Macdonald, B.M.; McArthur, S.M.; Zhou, M.; van Leeuwen, S. Refining expectations for environmental characteristics of refugia: Two ranges of differing elevation and topographical complexity are mesic refugia in an arid landscape. J. Biogeogr. 2017, 44, 2539–2550. [Google Scholar] [CrossRef]
- Byrne, M.; Coates, D.J.; Macdonald, B.M.; Hankinson, M.; McArthur, S.; van Leeuwen, S. High nuclear genetic differentiation, but low chloroplast diversity in a rare species, Aluta quadrata (Myrtaceae), with a disjunct distribution in the Pilbara, Western Australia. Aust. J. Bot. 2016, 64, 687–695. [Google Scholar] [CrossRef]
- Pepper, M.; Doughty, P.; Keogh, J.S. Geodiversity and endemism in the iconic Australian Pilbara region: A review of landscape evolution and biotic response in an ancient refugium. J. Biogeogr. 2013, 40, 1225–1239. [Google Scholar] [CrossRef]
- Gale, S. Long-term landscape evolution in Australia. Earth Surf. Process. Landf. 1992, 17, 323–343. [Google Scholar] [CrossRef]
- Cowling, R.M.; Potts, A.J.; Bradshaw, P.L.; Colville, J.; Arianoutsou, M.; Ferrier, S.; Forest, F.; Fyllas, N.M.; Hopper, S.D.; Ojeda, F.; et al. Variation in plant diversity in mediterranean-climate ecosystems: The role of climatic and topographical stability. J. Biogeogr. 2015, 42, 552–564. [Google Scholar] [CrossRef]
- Hopper, S.D. OCBIL theory: Towards an integrated understanding of the evolution, ecology and conservation of biodiversity on old, climatically buffered, infertile landscapes. Plant. Soil 2009, 322, 49–86. [Google Scholar] [CrossRef]
- Baldwin, B.G. Origins of plant diversity in the California Floristic Province. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 347–369. [Google Scholar] [CrossRef] [Green Version]
- Erickson, T.E.; Merritt, D.J. Introduction to plant diversity of the Pilbara. In Pilbara Seed Atlas and Field Guide; Erickson, T.E., Barrett, R.L., Merritt, D.J., Dixon, K.W., Eds.; CSIRO Publishing: Clayton South, Australia, 2016; pp. 1–5. [Google Scholar]
- Leighton, K.A. Climate. In Technical Bulletin No. 92: An Inventory and Condition Survey of the Pilbara Region, Western Australia; Van Vreeswyk, A., Payne, A., Leighton, K., Hennig, P., Eds.; Department of Agriculture, Government of Western Australia: Perth, Australia, 2004; pp. 19–38. [Google Scholar]
- Hill, K.D.; Johnson, L.A.S. Systematic studies in the eucalypts 7. A revision of the bloodwoods, genus Corymbia (Myrtaceae). Telopea 1995, 6, 185–505. [Google Scholar] [CrossRef]
- Wangka Maya Pilbara Aboriginal Language Centre. Anon Ngarluma Dictionary: English-Ngarluma Wordlist and Topical Wordlist; Wangka Maya Pilbara Aboriginal Language Centre: Roebourne, Australia.
- Tindale, M.D. Acacia Pruinocarpa, Flora of Australia, 11A: 181; ABRS/CSIRO Publishing: Melbourne, Australia, 2001. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Byrne, M.; Hankinson, M. Testing the variability of chloroplast sequences for plant phylogeography. Aust. J. Bot. 2012, 60, 569–574. [Google Scholar] [CrossRef]
- Gardner, M.G.; Fitch, A.J.; Bertozzi, T.; Lowe, A.J. Rise of the machines-recommendations for ecologists when using next generation sequencing for microsatellite development. Mol. Ecol. Resour. 2011, 11, 1093–1101. [Google Scholar] [CrossRef]
- Meglécz, E.; Costedoat, C.; Dubut, V.; Gilles, A.; Malausa, T.; Pech, N.; Martin, J.F. QDD: A user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics 2009, 26, 403–404. [Google Scholar] [CrossRef] [Green Version]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecuminism. Heredity (Edinb) 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rice, W.R. Analyzing tables of statistical tests. Evolution (N. Y.) 1989, 43, 223–225. [Google Scholar]
- Chapuis, M.P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. 1977, 39, 1–38. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the analysis of population structure. Evolution (N. Y.) 1984, 38, 1358–1370. [Google Scholar]
- Kolde, R. pheatmap: Pretty Heatmaps. R Package Version 1.0.10. Available online: https://cran.r-project.org/package=pheatmap (accessed on 28 March 2020).
- Slatkin, M. A measure of population subdivision based on microsatellite allele frequencies. Genetics 1995, 139, 457–462. [Google Scholar]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure: Extensions to linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faircloth, B.C.; Glenn, T.C. Protocol: Preparation of an AMPure XP Substitute (AKA Serapure); Cold Spring Harbor Protocols: New York, NY, USA, 2012; Web Document. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. 1999, 41, 95–98. [Google Scholar]
- Clarke, L.A.; Rebelo, C.S.; Gonçalves, J.; Boavida, M.G.; Jordan, P. PCR amplification introduces errors into mononucleotide and dinucleotide repeat sequences. J. Clin. Pathol. Mol. Pathol. 2001, 54, 351–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Ramos-Onsins, S.E.; Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Pons, O.; Petit, R.J. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [PubMed]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Polzin, T.; Daneschmand, S. V On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 2003, 31, 12–20. [Google Scholar] [CrossRef]
- Ennos, R.A. Estimating the relative rates of pollen and seed migration among plant populations. Heredity (Edinb) 1994, 72, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Crisp, M.D.; Burrows, G.E.; Cook, L.G.; Thornhill, A.H.; Bowman, D.M.J.S. Flammable biomes dominated by eucalypts originated at the Cretaceous-Palaeogene boundary. Nat. Commun. 2011, 2, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Heled, J.; Drummond, A.J. Calibrated tree priors for relaxed phylogenetics and divergence time estimation. Syst. Biol. 2012, 61, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Thornhill, A.H.; Popple, L.W.; Carter, R.J.; Ho, S.Y.W.; Crisp, M.D. Are pollen fossils useful for calibrating relaxed molecular clock dating of phylogenies? A comparative study using Myrtaceae. Mol. Phylogenet. Evol. 2012, 63, 15–27. [Google Scholar] [CrossRef]
- Gernhard, T. The conditioned reconstructed process. J. Theor. Biol. 2008, 21, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing Europe PMC Funders Group. Nat. Methods 2015, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.M.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermingham, E.; Moritz, C. Comparative phylogeography: Concepts and applications. Mol. Ecol. 1998, 367–369. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Keppel, G.; Van Niel, K.P.; Wardell-Johnson, G.W.; Yates, C.; Byrne, M.; Mucina, L.; Schut, A.G.T.; Hopper, S.D.; Franklin, S.E. Refugia: Identifying and understanding safe havens for biodiversity under climate change. Glob. Ecol. Biogeogr. 2012, 21, 393–404. [Google Scholar] [CrossRef]
- Lancaster, L.T.; Kay, K.M. Origin and diversification of the California flora: Re-examining classic hypotheses with molecular phylogenies. Evolution (N. Y.) 2013, 67, 1041–1054. [Google Scholar] [CrossRef]
- Abrams, K.M.; Huey, J.A.; Hillyer, M.J.; Humphreys, W.F.; Didham, R.K.; Harvey, M.S. Too hot to handle: Cenozoic aridification drives multiple independent incursions of Schizomida (Hubbardiidae) into hypogean environments. Mol. Phylogenet. Evol. 2019, 139, 106532. [Google Scholar] [CrossRef]
- Martin, H.A. Cenozoic climatic change and the development of the arid vegetation in Australia. J. Arid Environ. 2006, 66, 533–563. [Google Scholar] [CrossRef]
- Groeneveld, J.; Henderiks, J.; Renema, W.; McHugh, C.M.; De Vleeschouwer, D.; Christensen, B.A.; Fulthorpe, C.S.; Reuning, L.; Gallagher, S.J.; Bogus, K.; et al. Australian shelf sediments reveal shifts in Miocene Southern Hemisphere westerlies. Sci. Adv. 2017, 3, e1602567. [Google Scholar] [CrossRef] [Green Version]
- Crisp, M.D.; Isagi, Y.; Kato, Y.; Cook, L.G.; Bowman, D.M.J.S. Livistona palms in Australia: Ancient relics or opportunistic immigrants? Mol. Phylogenet. Evol. 2010, 54, 512–523. [Google Scholar] [CrossRef]
- Melville, J.; Ritchie, E.G.; Chapple, S.N.J.; Glor, R.E.; Schulte, J.A. Evolutionary origins and diversification of dragon lizards in Australia’s tropical savannas. Mol. Phylogenet. Evol. 2011, 58, 257–270. [Google Scholar] [CrossRef]
- Oliver, P.M.; Bauer, A.M. Systematics and evolution of the Australian knob-tail geckos (Nephrurus, Carphodactylidae, Gekkota): Plesiomorphic grades and biome shifts through the Miocene. Mol. Phylogenet. Evol. 2011, 59, 664–674. [Google Scholar] [CrossRef]
- Oliver, P.M.; Adams, M.; Doughty, P. Molecular evidence for ten species and Oligo-Miocene vicariance within a nominal Australian gecko species (Crenadactylus ocellatus, Diplodactylidae). BMC Evol. Biol. 2010, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pepper, M.; Doughty, P.; Arculus, R.; Keogh, J.S. Landforms predict phylogenetic structure on one of the world’s most ancient surfaces. BMC Evol. Biol. 2008, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M.; MacDonald, B.; Coates, D.J. Phylogeographical patterns in chloroplast DNA variation within the Acacia acuminata (Leguminosae: Mimosoideae) complex in Western Australia. J. Evol. Biol. 2002, 15, 576–587. [Google Scholar] [CrossRef]
- Byrne, M.; Hines, B. Phylogeographical analysis of cpDNA variation in Eucalyptus loxophleba (Myrtaceae). Aust. J. Bot. 2004, 52, 459–470. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Byrne, M. Congruence between phylogeographic patterns in cpDNA variation in Eucalyptus marginata (Myrtaceae) and geomorphology of the Darling Plateau, south-west of Western Australia. Aust. J. Bot. 2006, 54, 17–26. [Google Scholar] [CrossRef]
- Potts, A.J.; Hedderson, T.A.; Vlok, J.H.J.; Cowling, R.M. Pleistocene range dynamics in the eastern Greater Cape Floristic Region: A case study of the Little Karoo endemic Berkheya cuneata (Asteraceae). S. Afr. J. Bot. 2013, 88, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Steane, D.; M, B.; Vaillancourt, R.; Potts, B. Chloroplast DNA polymorphism signals complex interspecific interactions in Eucalyptus (Myrtaceae). Aust. Syst. Bot. 1998, 11, 25–40. [Google Scholar] [CrossRef]
- Freeman, J.; Jackson, H.; Steane, D.; McKinnon, G.; Dutkowski, G.; Potts, B.; Vaillancourt, R. Chloroplast DNA phylogeography of Eucalyptus globulus. Aust. J. Bot. 2001, 49, 585–596. [Google Scholar] [CrossRef]
- Jackson, H.D.; Steane, D.A.; Potts, B.M.; Vaillancourt, R.E. Chloroplast DNA evidence for reticulate evolution in Eucalyptus (Myrtaceae). Mol. Ecol. 1999, 8, 739–751. [Google Scholar] [CrossRef]
- McKinnon, G.E.; Steane, D.A.; Potts, B.M.; Vaillancourt, R.E. Incongruence between chloroplast and species phylogenies in Eucalyptus subgenus Monocalyptus (Myrtaceae). Am. J. Bot. 1999, 86, 1038–1046. [Google Scholar] [CrossRef] [Green Version]
- Healey, A.; Lee, D.J.; Furtado, A.; Henry, R.J. Evidence of inter-sectional chloroplast capture in Corymbia among sections Torellianae and Maculatae. Aust. J. Bot. 2018, 66, 369. [Google Scholar] [CrossRef]
- Schuster, T.M.; Setaro, S.D.; Tibbits, J.F.G.; Batty, E.L.; Fowler, R.M.; McLay, T.G.B.; Wilcox, S.; Ades, P.K.; Bayly, M.J. Chloroplast variation is incongruent with classification of the Australian bloodwood eucalypts (genus Corymbia, family Myrtaceae). PLoS ONE 2018, 13, 1–28. [Google Scholar] [CrossRef]
- Pepper, M.; Fujita, M.K.; Moritz, C.; Keogh, J.S. Palaeoclimate change drove diversification among isolated mountain refugia in the Australian arid zone. Mol. Ecol. 2011, 20, 1529–1545. [Google Scholar] [CrossRef]
- McCauley, D.E. The use of chloroplast DNA polymorphism in studies of gene flow in plants. Trends Ecol. Evol. 1995, 10, 198–202. [Google Scholar] [CrossRef]
- Zhang, D.; Hewitt, G.M. Nuclear DNA analyses in genetic studies of populations: Practice, problems and prospects. Mol. Ecol. 2003, 12, 563–584. [Google Scholar] [CrossRef]
- Janes, J.K.; Miller, J.M.; Dupuis, J.R.; Malenfant, R.M.; Gorrell, J.C.; Cullingham, C.I.; Andrew, R.L. The K = 2 conundrum. Mol. Ecol. 2017, 26, 3594–3602. [Google Scholar] [CrossRef] [Green Version]
- Meirmans, P.G. The trouble with isolation by distance. Mol. Ecol. 2012, 21, 2839–2846. [Google Scholar] [CrossRef]
- Lesica, P.; Allendorf, F.W. When are peripheral populations valuable for conservation? Conserv. Biol. 1995, 9, 753–760. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F. Genetic drift and estimation of effective population size. Genetics 1981, 98, 625–640. [Google Scholar]
- Loveless, M.D.; Hamrick, J.L. Ecological determinants of genetic structure in plant populations. Annu. Rev. Ecol. Syst. 1984, 15, 65–95. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding and Genetic Resources; Brown, A.H.D., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinauer: Sunderland, MA, USA, 1990; pp. 43–63. [Google Scholar]
- Odee, D.W.; Telford, A.; Wilson, J.; Gaye, A.; Cavers, S. Plio-Pleistocene history and phylogeography of Acacia senegal in dry woodlands and savannahs of sub-Saharan tropical Africa: Evidence of early colonisation and recent range expansion. Heredity (Edinb) 2012, 109, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Ruiz Guajardo, J.C.; Schnabel, A.; Ennos, R.; Preuss, S.; Otero-Arnaiz, A.; Stone, G. Landscape genetics of the key African acacia species Senegalia mellifera (Vahl)- The importance of the Kenyan Rift Valley. Mol. Ecol. 2010, 19, 5126–5139. [Google Scholar] [CrossRef]
- Maddison, W.; Knowles, L. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 2006, 55, 21–30. [Google Scholar] [CrossRef]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevill, P.G.; Bradbury, D.; Williams, A.; Tomlinson, S.; Krauss, S.L. Genetic and palaeo-climatic evidence for widespread persistence of the coastal tree species Eucalyptus gomphocephala (Myrtaceae) during the Last Glacial Maximum. Ann. Bot. 2014, 113, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Nathan, R.; Schurr, F.M.; Spiegel, O.; Steinitz, O.; Trakhtenbrot, A.; Tsoar, A. Mechanisms of long-distance seed dispersal. Trends Ecol. Evol. 2008, 23, 638–647. [Google Scholar] [CrossRef]
- Visher, S.S. Tropical cyclones and the dispersal of life from island to island in the Pacific. Am. Nat. 1925, 59, 70–78. [Google Scholar] [CrossRef]
- Higgins, A.S.I.; Nathan, R.; Cain, M.L. Are Long-Distance Dispersal Events in Plants Usually Caused by Nonstandard Means of Dispersal? Ecology 2003, 84, 1945–1956. [Google Scholar] [CrossRef]
- VanVreeswyk, A.; Leighton, K.; Payne, A.; Hennig, P. An Inventory and Condition Survey of the Pilbara Region, Western Australia; Department of Agriculture and Food: Perth, Australia, 2004; Volume 92, pp. 19–38.
- Calviño-Cancela, A.M.; Dunn, R.R.; Etten, E.J.B.V.; Lamont, B.B.; Dunn, R.R.; Etten, E.J.B.V.; Lamont, B.B.; Calvifio-cancela, M. Emus as non-standard seed dispersers and their potential for long-distance dispersal. Ecography (Cop.) 2006, 29, 632–640. [Google Scholar] [CrossRef]
- Tobler, R.; Rohrlach, A.; Soubrier, J.; Bover, P.; Llamas, B.; Tuke, J.; Bean, N.; Abdullah-Highfold, A.; Agius, S.; O’Donoghue, A.; et al. Aboriginal mitogenomes reveal 50,000 years of regionalism in Australia. Nature 2017, 544, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Silcock, J.L. Aboriginal Translocations: The Intentional Propagation and Dispersal of Plants in Aboriginal Australia. J. Ethnobiol. 2018, 38, 390–405. [Google Scholar] [CrossRef]
- Bowman, D.M.J.S.; Gibson, J.; Kondo, T. Aboriginal myth meets DNA analysis. Nature 2015, 520, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetto, M.; Ens, E.J.; Honings, T.; Wilson, P.D.; Yap, J.Y.S.; Costello, O.; Round, E.R.; Bowern, C. From Songlines to genomes: Prehistoric assisted migration of a rain forest tree by Australian Aboriginal people. PLoS ONE 2017, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lullfitz, A.; Byrne, M.; Knapp, L.; Hopper, S.D. Platysace (Apiaceae) of south-western Australia: Silent story tellers of an ancient human landscape. Biol. J. Linn. Soc. 2020, 130, 61–78. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop | n | p | NA | NAP | HO | HE | FIS | Range |
|---|---|---|---|---|---|---|---|---|
| C. hamersleyana | ||||||||
| BEN | 24 | 85.71 | 5.57 (0.88) | 1 | 0.47 (0.08) | 0.58 (0.08) | 0.18 * | |
| COP | 24 | 100 | 6.11 (1.04) | 0.47 (0.08) | 0.58 (0.08) | 0.19 * | ||
| DAW | 24 | 100 | 6.63 (1.14) | 0.49 (0.07) | 0.60 (0.08) | 0.18 * | Chichester | |
| DEE | 24 | 100 | 6.52 (0.94) | 1 | 0.52 (0.06) | 0.63 (0.06) | 0.17 * | |
| DIN | 24 | 100 | 7.29 (1.04) | 3 | 0.47 (0.07) | 0.64 (0.06) | 0.26 * | Hamersley |
| EMU | 24 | 100 | 6.97 (1.04) | 2 | 0.51 (0.08) | 0.61 (0.08) | 0.15 * | Hamersley |
| GAL | 24 | 100 | 7.33 (1.24) | 0.42 (0.07) | 0.60 (0.09) | 0.29 * | ||
| GID | 24 | 100 | 7.65 (1.25) | 0.47 (0.07) | 0.62 (0.08) | 0.23 * | Hamersley | |
| HAM | 24 | 100 | 7.06 (1.26) | 0.52 (0.07) | 0.61 (0.08) | 0.14 * | Hamersley | |
| HOO | 24 | 100 | 6.61 (1.11) | 0.49 (0.08) | 0.57 (0.08) | 0.14 * | Chichester | |
| KAN | 24 | 100 | 6.84 (1.02) | 0.46 (0.07) | 0.59 (0.07) | 0.21 * | ||
| KAR | 24 | 100 | 7.00 (1.17) | 2 | 0.47 (0.07) | 0.60 (0.08) | 0.21 * | |
| MCK | 24 | 100 | 6.22 (1.10) | 3 | 0.49 (0.07) | 0.58 (0.07) | 0.15 * | Chichester |
| MIN | 24 | 100 | 6.54 (0.84) | 2 | 0.49 (0.08) | 0.59 (0.07) | 0.17 * | |
| NUL | 24 | 100 | 6.21 (0.91) | 1 | 0.46 (0.08) | 0.62 (0.08) | 0.25 * | |
| ORD | 24 | 92.86 | 6.68 (1.08) | 0.50 (0.07) | 0.62 (0.08) | 0.19 * | ||
| PAM | 24 | 92.86 | 7.08 (1.16) | 1 | 0.47 (0.08) | 0.59 (0.09) | 0.20 * | Hamersley |
| RUB | 24 | 92.86 | 6.40 (1.15) | 3 | 0.52 (0.08) | 0.59 (0.08) | 0.12 * | |
| SHA | 24 | 100 | 6.30 (0.98) | 1 | 0.51 (0.06) | 0.64 (0.07) | 0.19 * | |
| WEE | 24 | 100 | 6.44 (1.05) | 1 | 0.45 (0.08) | 0.60 (0.08) | 0.24 * | Hamersley |
| Mean (SE) | 24.00 (0) | 98.21 (0.88) | 6.67 (0.11) | 1.88 (0.27) | 0.48 (0.01) | 0.60 (0.01) | 0.19 (0.01) | |
| A. pruinocarpa | ||||||||
| ANG | 20 | 92.86 | 4.69 (0.71) | 0.42 (0.07) | 0.52 (0.07) | 0.20 * | Hamersley | |
| BUN | 24 | 100 | 4.55 (0.70) | 0.48 (0.05) | 0.58 (0.06) | 0.18 * | ||
| COO | 23 | 92.86 | 4.68 (0.71) | 1 | 0.41 (0.06) | 0.57 (0.07) | 0.29 * | |
| COR | 24 | 85.71 | 4.01 (0.66) | 2 | 0.46 (0.07) | 0.50 (0.07) | 0.08 * | Chichester |
| HAM | 24 | 92.86 | 4.33 (0.78) | 1 | 0.54 (0.07) | 0.55 (0.07) | 0.02 | Hamersley |
| HAR | 24 | 100 | 4.41 (0.73) | 1 | 0.45 (0.07) | 0.50 (0.07) | 0.11 * | Hamersley |
| HES | 23 | 92.86 | 4.86 (0.85) | 1 | 0.41 (0.06) | 0.51 (0.07) | 0.20 * | Chichester |
| HIL | 24 | 92.86 | 5.05 (0.91) | 1 | 0.45 (0.06) | 0.54 (0.07) | 0.18 * | Hamersley |
| HRP | 24 | 85.71 | 4.57 (0.95) | 0.40 (0.08) | 0.47 (0.08) | 0.15 * | Chichester | |
| MSJ | 23 | 92.86 | 4.49 (0.80) | 1 | 0.45 (0.07) | 0.54 (0.07) | 0.18 * | |
| MET | 22 | 92.86 | 4.90 (0.94) | 0.47 (0.07) | 0.56 (0.08) | 0.16 * | Hamersley | |
| NAM | 24 | 92.86 | 4.45 (0.78) | 1 | 0.42 (0.07) | 0.50 (0.08) | 0.17 * | Hamersley |
| OPH | 23 | 100 | 4.66 (0.64) | 3 | 0.41 (0.07) | 0.53 (0.07) | 0.24 * | Hamersley |
| OXE | 23 | 100 | 4.67 (0.77) | 0.43 (0.06) | 0.54 (0.07) | 0.20 * | Hamersley | |
| RHL | 20 | 92.86 | 4.06 (0.63) | 0.41 (0.07) | 0.50 (0.08) | 0.18 * | Chichester | |
| RHO | 23 | 92.86 | 3.87 (0.56) | 0.39 (0.07) | 0.46 (0.08) | 0.14 * | Hamersley | |
| RIO | 23 | 92.86 | 4.25 (0.78) | 1 | 0.43 (0.07) | 0.51 (0.07) | 0.17 * | Hamersley |
| SPE | 23 | 92.86 | 4.58 (0.90) | 1 | 0.42 (0.07) | 0.51 (0.07) | 0.17 * | Hamersley |
| STE | 24 | 100 | 4.18 (0.69) | 0.35 (0.07) | 0.47 (0.07) | 0.25 * | ||
| WAR | 24 | 100 | 4.56 (0.66) | 1 | 0.46 (0.07) | 0.55 (0.07) | 0.15 * | Hamersley |
| WRA | 24 | 92.86 | 4.80 (0.84) | 1 | 0.44 (0.08) | 0.47 (0.07) | 0.05 * | Hamersley |
| YAM | 24 | 85.71 | 4.58 (0.83) | 0.42 (0.07) | 0.49 (0.08) | 0.14 * | Hamersley | |
| YAR | 24 | 100 | 4.59 (0.76) | 0.42 (0.06) | 0.53 (0.07) | 0.21 * | Hamersley | |
| Mean (SE) | 23.22(0.24) | 94.10 (0.97) | 4.51 (0.06) | 1.23 (0.17) | 0.43 (0.01) | 0.52 (0.01) | 0.17 (0.01) | |
| Species | h | hd | π | Tajima’s D | Fu’s Fs | R2 | Demographic Expansion | Spatial Expansion | Pollen/Seed Flow Ratio |
|---|---|---|---|---|---|---|---|---|---|
| C. hamersleyana | 14 | 0.759 | 0.003 (0.00) | 0.607 ns | 2.474 ns | 0.086 * | SSD (p = 0.31):Hrag NS | SSD (p = 0.51):Hrag NS | 6.67 (6.45–6.89) |
| A. pruinocarpa | 37 | 0.903 | 0.002 (0.00) | −0.844 ns | −4.982 ns | 0.082 * | SSD (p = 0.42):Hrag NS | SSD (p = 0.49):Hrag NS | 2.96 (2.69–3.23 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nistelberger, H.M.; Binks, R.M.; van Leeuwen, S.; Coates, D.J.; McArthur, S.L.; Macdonald, B.M.; Hankinson, M.; Byrne, M. Extensive Genetic Connectivity and Historical Persistence Are Features of Two Widespread Tree Species in the Ancient Pilbara Region of Western Australia. Genes 2020, 11, 863. https://doi.org/10.3390/genes11080863
Nistelberger HM, Binks RM, van Leeuwen S, Coates DJ, McArthur SL, Macdonald BM, Hankinson M, Byrne M. Extensive Genetic Connectivity and Historical Persistence Are Features of Two Widespread Tree Species in the Ancient Pilbara Region of Western Australia. Genes. 2020; 11(8):863. https://doi.org/10.3390/genes11080863
Chicago/Turabian StyleNistelberger, Heidi M., Rachel M. Binks, Stephen van Leeuwen, David J. Coates, Shelley L. McArthur, Bronwyn M. Macdonald, Margaret Hankinson, and Margaret Byrne. 2020. "Extensive Genetic Connectivity and Historical Persistence Are Features of Two Widespread Tree Species in the Ancient Pilbara Region of Western Australia" Genes 11, no. 8: 863. https://doi.org/10.3390/genes11080863
APA StyleNistelberger, H. M., Binks, R. M., van Leeuwen, S., Coates, D. J., McArthur, S. L., Macdonald, B. M., Hankinson, M., & Byrne, M. (2020). Extensive Genetic Connectivity and Historical Persistence Are Features of Two Widespread Tree Species in the Ancient Pilbara Region of Western Australia. Genes, 11(8), 863. https://doi.org/10.3390/genes11080863