Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Data Sets
2.2. Analysis Framework
2.3. Extraction of the Candidate Genes
3. Results
3.1. Single-SNP Based GWAS Analysis
3.2. Detection of Genotype-Phenotype Association Using the Combined Framework
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.C.; Nelson, G.W.; Troyer, J.L.; Lautenberger, J.A.; Kessing, B.D.; Winkler, C.A.; O’Brien, S.J. Accounting for multiple comparisons in a genome-wide association study (GWAS). BMC Genom. 2010, 11, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, W.S.; Moore, J.H. Genome-wide association studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Holland, D.; Fan, C.C.; Frei, O.; Shadrin, A.A.; Smeland, O.B.; Sundar, V.; Andreassen, O.A.; Dale, A.M. Estimating inflation in GWAS summary statistics due to variance distortion from cryptic relatedness. BioRxiv 2017, 164939. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Mao, Y.; Xie, C.; Smith, H.; Luo, L.; Xu, S. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics 2005, 169, 2267–2275. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821. [Google Scholar] [CrossRef] [Green Version]
- Eu-Ahsunthornwattana, J.; Miller, E.N.; Fakiola, M.; Jeronimo, S.M.; Blackwell, J.M.; Cordell, H.J.; Wellcome Trust Case Control Consortium 2. Comparison of methods to account for relatedness in genome-wide association studies with family-based data. PLoS Genet. 2014, 10, e1004445. [Google Scholar] [CrossRef] [PubMed]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Young, A.I. Solving the missing heritability problem. PLoS Genet. 2019, 15, e1008222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.D.; Langley, C.H. The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res. 1999, 9, 720–731. [Google Scholar]
- Akey, J.; Jin, L.; Xiong, M. Haplotypes vs single marker linkage disequilibrium tests: What do we gain? Eur. J. Hum. Genet. 2001, 9, 291. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Calabrese, P.; Nordborg, M.; Sun, F. Haplotype block structure and its applications to association studies: Power and study designs. Am. J. Hum. Genet. 2002, 71, 1386–1394. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, A.J.; Hamblin, M.T.; Jannink, J.L. Performance of single nucleotide polymorphisms versus haplotypes for genome-wide association analysis in barley. PLoS ONE 2010, 5, e14079. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Bennewitz, J. Invited review: Genome-wide association analysis for quantitative traits in livestock—A selective review of statistical models and experimental designs. Arch. Tierz. 2017, 60, 335. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Jia, Z.; Dunwell, J.M. The applications of new multi-locus GWAS methodologies in the genetic dissection of complex traits. Front. Plant Sci. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Zhang, F.; Zhou, Y. The application of multi-Locus GWAS for the detection of salt-tolerance loci in rice. Front. Plant Sci. 2018, 9, 1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Liu, M.; Yan, Y.; Qing, C.; Zhang, X.; Zhang, Y.; Long, Y.; Wang, L.; Pan, L.; Zou, C.; et al. Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yang, T.; Zhou, Y.; Yin, S.; Li, P.; Liu, J.; Xu, S.; Yang, Z.; Xu, C. Genome-wide association mapping of starch pasting properties in maize using single-locus and multi-locus models. Front. Plant Sci. 2018, 9, 1311. [Google Scholar] [CrossRef]
- Abed, A.; Belzile, F. Comparing Single-SNP, Multi-SNP, and Haplotype-Based Approaches in Association Studies for Major Traits in Barley. Plant Genome 2019, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, F.; Zhai, R.; Lin, X.; Wang, Z.; Su, L.; Christiani, D.C. Correction for population stratification in random forest analysis. Int. J. Epidemiol. 2012, 41, 1798–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.; Huang, J.Z.; Wu, Q.; Nguyen, T.T.; Li, M.J. Genome-wide association data classification and SNPs selection using two-stage quality-based Random Forests. BMC Genom. 2015, 16, 5. [Google Scholar] [CrossRef] [Green Version]
- Armero, C.; Cabras, S.; Castellanos, M.E.; Quirós, A. Two-Stage Bayesian Approach for GWAS With Known Genealogy. J. Comput. Graph. Stat. 2019, 28, 197–204. [Google Scholar] [CrossRef]
- Huang, X.; Zhou, W.; Bellis, E.S.; Stubblefield, J.; Causey, J.; Qualls, J.; Walker, K. Minor QTLs mining through the combination of GWAS and machine learning feature selection. BioRxiv 2019, 712190. [Google Scholar] [CrossRef]
- Brieuc, M.S.; Waters, C.D.; Drinan, D.P.; Naish, K.A. A practical introduction to Random Forest for genetic association studies in ecology and evolution. Mol. Ecol. Resour. 2018, 18, 755–766. [Google Scholar] [CrossRef]
- Ramzan, F.; Klees, S.; Schmitt, A.O.; Cavero, D.; Gültas, M. Identification of Age-Specific and Common Key Regulatory Mechanisms Governing Eggshell Strength in Chicken Using Random Forests. Genes 2020, 11, 464. [Google Scholar] [CrossRef]
- Romagnoni, A.; Jégou, S.; Van Steen, K.; Wainrib, G.; Hugot, J.P. Comparative performances of machine learning methods for classifying Crohn Disease patients using genome-wide genotyping data. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Van der Heide, E.; Veerkamp, R.; van Pelt, M.; Kamphuis, C.; Athanasiadis, I.; Ducro, B. Comparing regression, naive Bayes, and random forest methods in the prediction of individual survival to second lactation in Holstein cattle. J. Dairy Sci. 2019, 102, 9409–9421. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, N.; Wang, Y.G.; George, A.W.; Reverter, A.; Li, Y. Genomic prediction of breeding values using a subset of SNPs identified by three machine learning methods. Front. Genet. 2018, 9, 237. [Google Scholar] [CrossRef]
- Hamblin, M.T.; Jannink, J.L. Factors affecting the power of haplotype markers in association studies. Plant Genome 2011, 4, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Sarti, F.; Lasagna, E.; Ceccobelli, S.; Di Lorenzo, P.; Filippini, F.; Sbarra, F.; Giontella, A.; Pieramati, C.; Panella, F. Influence of single nucleotide polymorphisms in the myostatin and myogenic factor 5 muscle growth-related genes on the performance traits of Marchigiana beef cattle. J. Anim. Sci. 2014, 92, 3804–3810. [Google Scholar] [CrossRef] [PubMed]
- Sarti, F.M.; Ceccobelli, S.; Lasagna, E.; Di Lorenzo, P.; Sbarra, F.; Pieramati, C.; Giontella, A.; Panella, F. Influence of single nucleotide polymorphisms in some candidate genes related to the performance traits in Italian beef cattle breeds. Livest. Sci. 2019, 230, 103834. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, L.; Wu, X.; Li, B.; Huang, W.; Weng, Z.; Lin, Z.; Song, L.; Guo, Y.; Meng, Z.; et al. Identification of Candidate Growth-Related SNPs and Genes Using GWAS in Brown-Marbled Grouper (Epinephelus fuscoguttatus). Mar. Biotechnol. 2020, 22, 153–166. [Google Scholar] [CrossRef]
- Freebern, E.; Santos, D.J.; Fang, L.; Jiang, J.; Gaddis, K.L.P.; Liu, G.E.; Vanraden, P.M.; Maltecca, C.; Cole, J.B.; Ma, L. GWAS and fine-mapping of livability and six disease traits in Holstein cattle. BMC Genom. 2020, 21, 41. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.P.; Guatteo, R.; Davergne, A.; Saout, J.; Grohs, C.; Deloche, M.C.; Taussat, S.; Fritz, S.; Boussaha, M.; Blanquefort, P.; et al. Identification of the ABCC4, IER3, and CBFA2T2 candidate genes for resistance to paratuberculosis from sequence-based GWAS in Holstein and Normande dairy cattle. Genet. Sel. Evol. 2020, 52, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Sinclair-Waters, M.; Ødegård, J.; Korsvoll, S.A.; Moen, T.; Lien, S.; Primmer, C.R.; Barson, N.J. Beyond large-effect loci: Large-scale GWAS reveals a mixed large-effect and polygenic architecture for age at maturity of Atlantic salmon. Genet. Sel. Evol. 2020, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.S.; Ruyter, B.; Meuwissen, T.H.; Moghadam, H.; Hillestad, B.; Sonesson, A.K. GWAS identifies genetic variants associated with omega-3 fatty acid composition of Atlantic salmon fillets. Aquaculture 2020, 514, 734494. [Google Scholar] [CrossRef]
- Nicholls, H.L.; John, C.R.; Watson, D.S.; Munroe, P.B.; Barnes, M.R.; Cabrera, C.P. Reaching the End-Game for GWAS: Machine Learning Approaches for the Prioritization of Complex Disease Loci. Front. Genet. 2020, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Misra, G.; Badoni, S.; Anacleto, R.; Graner, A.; Alexandrov, N.; Sreenivasulu, N. Whole genome sequencing-based association study to unravel genetic architecture of cooked grain width and length traits in rice. Sci. Rep. 2017, 7, 1–16. [Google Scholar]
- Li, C.; Fu, Y.; Sun, R.; Wang, Y.; Wang, Q. Single-locus and multi-locus genome-wide association studies in the genetic dissection of fiber quality traits in upland cotton (Gossypium hirsutum L.). Front. Plant Sci. 2018, 9, 1083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Roeder, K.; Wallstrom, G.; Devlin, B. Integration of association statistics over genomic regions using Bayesian adaptive regression splines. Hum. Genom. 2003, 1, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, D.F.; Szymczak, S.; Ziegler, A.; König, I.R. Picking single-nucleotide polymorphisms in forests. BMC Proc. 2007, 1, 59. [Google Scholar] [CrossRef] [Green Version]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, C.; Yan, Y.; Li, G.; Wu, G.; Liu, A.; Yang, N. Genome-wide association analysis of age-dependent egg weights in chickens. Front. Genet. 2018, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Lippert, C.; Listgarten, J.; Liu, Y.; Kadie, C.M.; Davidson, R.I.; Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 2011, 8, 833. [Google Scholar] [CrossRef]
- Gao, X.; Becker, L.C.; Becker, D.M.; Starmer, J.D.; Province, M.A. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2010, 34, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.N. Generalized Additive Models: An Introduction with R; Chapman and Hall/CRC: Boca Raton, FL, USA, 2017. [Google Scholar]
- Silverman, B.W. Some aspects of the spline smoothing approach to non-parametric regression curve fitting. J. R. Stat. Soc. Ser. B (Methodol.) 1985, 47, 1–21. [Google Scholar] [CrossRef]
- Beissinger, T.M.; Rosa, G.J.; Kaeppler, S.M.; Gianola, D.; De Leon, N. Defining window-boundaries for genomic analyses using smoothing spline techniques. Genet. Sel. Evol. 2015, 47, 30. [Google Scholar] [CrossRef] [Green Version]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the Boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Ozawa, H.; Nagai, H. Histochemical studies of Ca-ATPase, succinate and NAD+-dependent isocitrate dehydrogenases in the shell gland of laying Japanese quails: With special reference to calcium-transporting cells. Histochemistry 1985, 83, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, F.; Qu, H.; Luo, C.; Wang, J.; Shu, D. Associations between variants of bone morphogenetic protein 7 gene and growth traits in chickens. Br. Poult. Sci. 2018, 59, 264–269. [Google Scholar] [CrossRef]
- Jin, S. Bipotent stem cells support the cyclical regeneration of endometrial epithelium of the murine uterus. Proc. Natl. Acad. Sci. USA 2019, 116, 6848–6857. [Google Scholar] [CrossRef] [Green Version]
- Boyle, M.; Wong, C.; Rocha, M.; Jones, D.L. Decline in self-renewal factors contributes to aging of the stem cell niche in the Drosophila testis. Cell Stem Cell 2007, 1, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.L.; Chen, Y.T.; Smith, S.J.; James Nelson, W. Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin–green fluorescent protein. J. Cell Biol. 1998, 142, 1105–1119. [Google Scholar] [CrossRef]
- Dahl, U.; Sjödin, A.; Larue, L.; Radice, G.L.; Cajander, S.; Takeichi, M.; Kemler, R.; Semb, H. Genetic dissection of cadherin function during nephrogenesis. Mol. Cell. Biol. 2002, 22, 1474–1487. [Google Scholar] [CrossRef] [Green Version]
- Marie, P.J.; Haÿ, E.; Modrowski, D.; Revollo, L.; Mbalaviele, G.; Civitelli, R. Cadherin-mediated cell–cell adhesion and signaling in the skeleton. Calcif. Tissue Int. 2014, 94, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Levin, M.H.; Marín-Briggiler, C.I.; Caballero, J.N.; Veiga, M.F. Epithelial and neural cadherin expression in the mammalian reproductive tract and gametes and their participation in fertilization-related events. Dev. Biol. 2015, 401, 2–16. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.F.; Hou, Z.C.; Yi, G.Q.; Xu, G.Y.; Yang, N. The sodium channel gene family is specifically expressed in hen uterus and associated with eggshell quality traits. BMC Genet. 2013, 14, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleet, J.C.; Schoch, R.D. Molecular mechanisms for regulation of intestinal calcium absorption by vitamin D and other factors. Crit. Rev. Clin. Lab. Sci. 2010, 47, 181–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josephs, E.B.; Stinchcombe, J.R.; Wright, S.I. What can genome-wide association studies tell us about the evolutionary forces maintaining genetic variation for quantitative traits? New Phytol. 2017, 214, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, D.; He, F.; Wang, J.; Joshi, T.; Xu, D. Phenotype prediction and genome-wide association study using deep convolutional neural network of soybean. Front. Genet. 2019, 10, 1091. [Google Scholar] [CrossRef]
- Zaykin, D.V.; Zhivotovsky, L.A.; Westfall, P.H.; Weir, B.S. Truncated product method for combining P-values. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2002, 22, 170–185. [Google Scholar]
- Dudbridge, F.; Koeleman, B.P. Rank truncated product of P-values, with application to genomewide association scans. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2003, 25, 360–366. [Google Scholar] [CrossRef]
- Yang, H.C.; Lin, C.Y.; Fann, C.S. A sliding-window weighted linkage disequilibrium test. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2006, 30, 531–545. [Google Scholar] [CrossRef]
- Yang, H.C.; Hsieh, H.Y.; Fann, C.S. Kernel-based association test. Genetics 2008, 179, 1057–1068. [Google Scholar] [CrossRef] [Green Version]
- Brionne, A.; Nys, Y.; Hennequet-Antier, C.; Gautron, J. Hen uterine gene expression profiling during eggshell formation reveals putative proteins involved in the supply of minerals or in the shell mineralization process. BMC Genom. 2014, 15, 220. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.M.; Cutkomp, L. In vitro studies of DDT, DDE, and ATPase as related to avian eggshell thinning. Arch. Environ. Contam. Toxicol. 1982, 11, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Jonchère, V.; Brionne, A.; Gautron, J.; Nys, Y. Identification of uterine ion transporters for mineralisation precursors of the avian eggshell. BMC Physiol. 2012, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Chakraborti, S.; Dhalla, N.S. Regulation of Membrane Na+-K+ ATPase; Springer: Berlin, Germany, 2016. [Google Scholar]
- Morth, J.P.; Pedersen, B.P.; Buch-Pedersen, M.J.; Andersen, J.P.; Vilsen, B.; Palmgren, M.G.; Nissen, P. A structural overview of the plasma membrane Na+, K+-ATPase and H+-ATPase ion pumps. Nat. Rev. Mol. Cell Biol. 2011, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Jin, S.; Ma, C.; Wang, Z.; Fang, Q.; Jiang, R. RNA-Seq reveals seven promising candidate genes affecting the proportion of thick egg albumen in layer-type chickens. Sci. Rep. 2017, 7, 1–9. [Google Scholar]
- Yin, Z.; Lian, L.; Zhu, F.; Zhang, Z.H.; Hincke, M.; Yang, N.; Hou, Z.C. The transcriptome landscapes of ovary and three oviduct segments during chicken (Gallus gallus) egg formation. Genomics 2020, 112, 243–251. [Google Scholar] [CrossRef]
- Elks, C.E.; Den Hoed, M.; Zhao, J.H.; Sharp, S.J.; Wareham, N.J.; Loos, R.J.; Ong, K.K. Variability in the heritability of body mass index: A systematic review and meta-regression. Front. Endocrinol. 2012, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Sillanpää, M.J.; Silventoinen, K.; Kaprio, J.; Pitkäniemi, J. Estimating modifying effect of age on genetic and environmental variance components in twin models. Genetics 2016, 202, 1313–1328. [Google Scholar] [CrossRef] [Green Version]





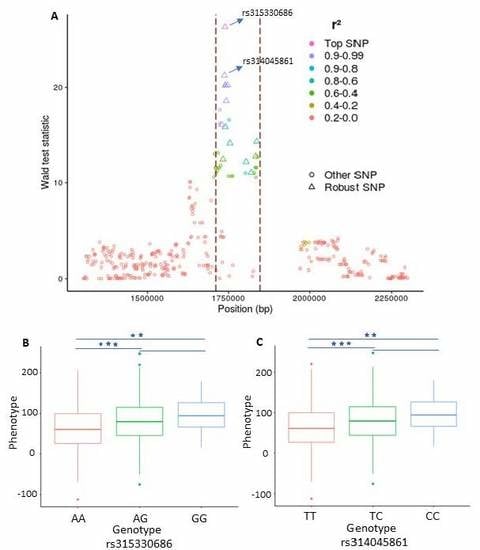
{kind=link}
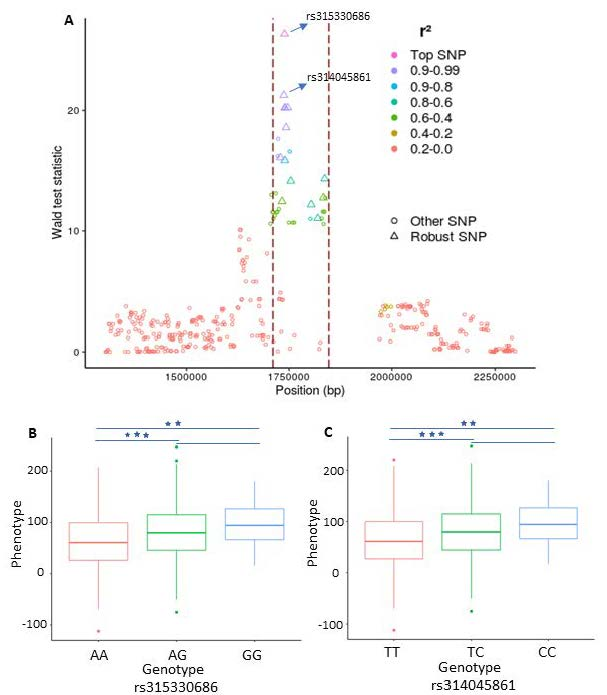
{kind=link}
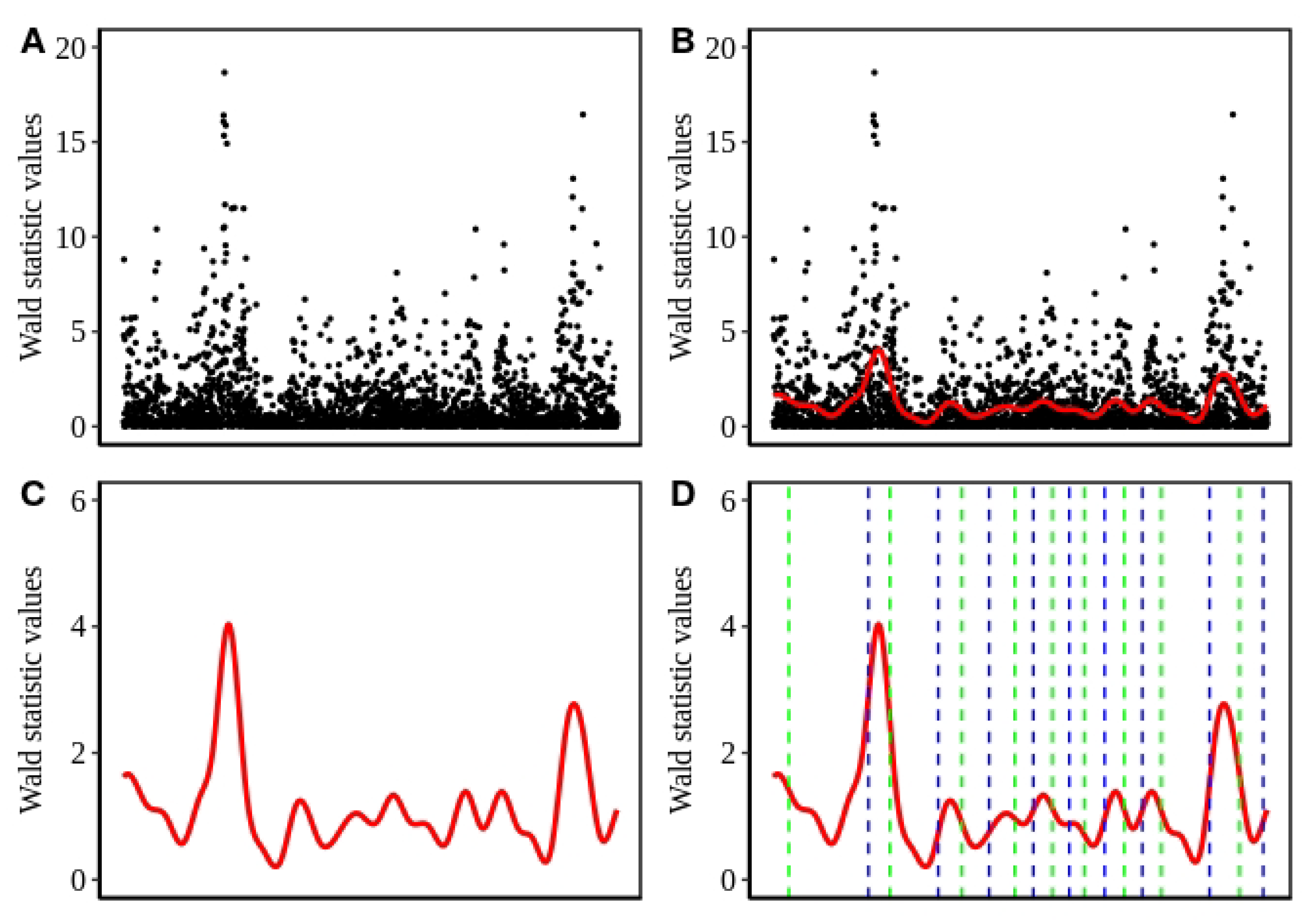
{kind=link}
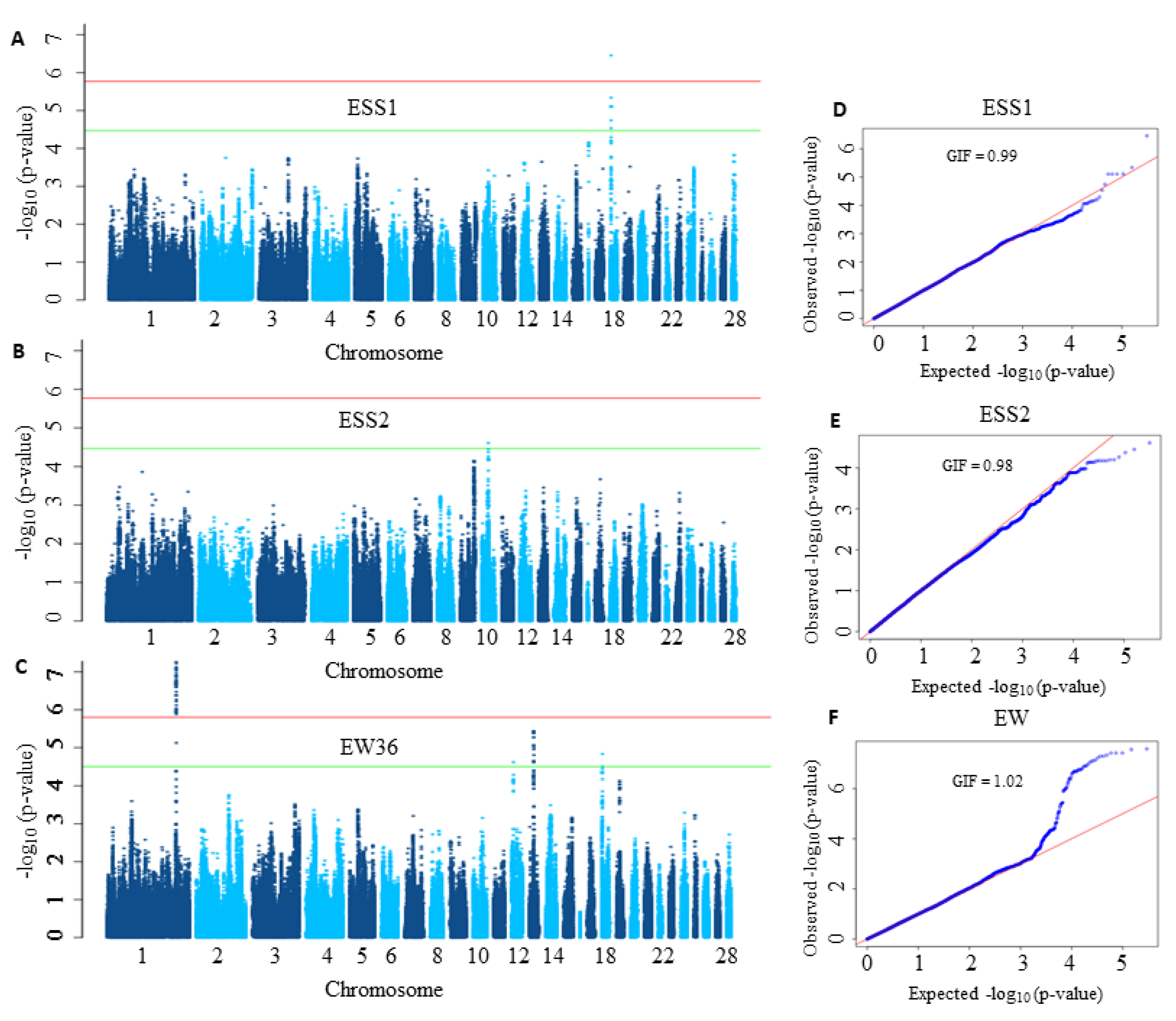
{kind=link}
{kind=link}
| Chromosome | No. of SNPs | Start Position | End Position | No. of Genes | Trait |
|---|---|---|---|---|---|
| 2 | 204 | 147,575,318 | 148,273,465 | 3 | ESS1 |
| 9 | 66 | 21,762,694 | 21,953,310 | 0 | ESS1 |
| 9 | 82 | 21,777,888 | 22,001,729 | 0 | ESS2 |
| 10 | 75 | 6,517,673 | 6,728,897 | 4 | ESS1 |
| 10 | 86 | 9,922,422 | 10,054,824 | 2 | ESS1 |
| 10 | 60 | 10,715,120 | 10,818,097 | 3 | ESS2 |
| 10 | 61 | 11,245,585 | 11,351,799 | 1 | ESS2 |
| 12 | 112 | 10,948,518 | 11,227,521 | 2 | ESS1 |
| 15 | 42 | 4,908,007 | 5,006,688 | 7 | ESS1 |
| 15 | 43 | 6,193,090 | 6,273,778 | 3 | ESS2 |
| 18 | 38 | 1,722,586 | 1,836,741 | 2 | ESS1 |
| 20 | 51 | 7,589,607 | 7,717,177 | 1 | ESS1 |
| 20 | 46 | 7,599,368 | 7,711,505 | 1 | ESS2 |
| Chromosome | No. of SNPs | Start Position | End Position | No. of Genes |
|---|---|---|---|---|
| 1 | 304 | 167,931,038 | 169,505,140 | 25 |
| 4 | 205 | 17,189,770 | 18,080,445 | 9 |
| 4 | 143 | 21,319,808 | 21,849,558 | 3 |
| 4 | 136 | 77,317,446 | 78,081,369 | 4 |
| 12 | 39 | 2,849,562 | 3,010,032 | 7 |
| 13 | 49 | 8,495,533 | 8,608,578 | 6 |
| 14 | 58 | 7,023,793 | 7,188,250 | 4 |
| 15 | 41 | 11,193,342 | 11,309,808 | 8 |
| 15 | 35 | 11,419,957 | 11,514,516 | 3 |
| 18 | 30 | 1,057,714 | 1,136,220 | 1 |
| 18 | 28 | 1,179,899 | 1,238,583 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramzan, F.; Gültas, M.; Bertram, H.; Cavero, D.; Schmitt, A.O. Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations. Genes 2020, 11, 892. https://doi.org/10.3390/genes11080892
Ramzan F, Gültas M, Bertram H, Cavero D, Schmitt AO. Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations. Genes. 2020; 11(8):892. https://doi.org/10.3390/genes11080892
Chicago/Turabian StyleRamzan, Faisal, Mehmet Gültas, Hendrik Bertram, David Cavero, and Armin Otto Schmitt. 2020. "Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations" Genes 11, no. 8: 892. https://doi.org/10.3390/genes11080892
APA StyleRamzan, F., Gültas, M., Bertram, H., Cavero, D., & Schmitt, A. O. (2020). Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations. Genes, 11(8), 892. https://doi.org/10.3390/genes11080892