A Four-Methylated lncRNAs-Based Prognostic Signature for Hepatocellular Carcinoma


Abstract
1. Introduction
2. Materials and Methods
2.1. Data and Sources
2.2. Identification of Differential Expressed lncRNAs with Altered Methylation Status in HCC
2.3. Establishment of Prognostic Predictive Model and ROC Curve
2.4. LncRNAs Survival Analysis
2.5. Use Prognostic Risk Score Model to Evaluate HCC in Cell Line
2.6. Statistical Analyses
3. Results
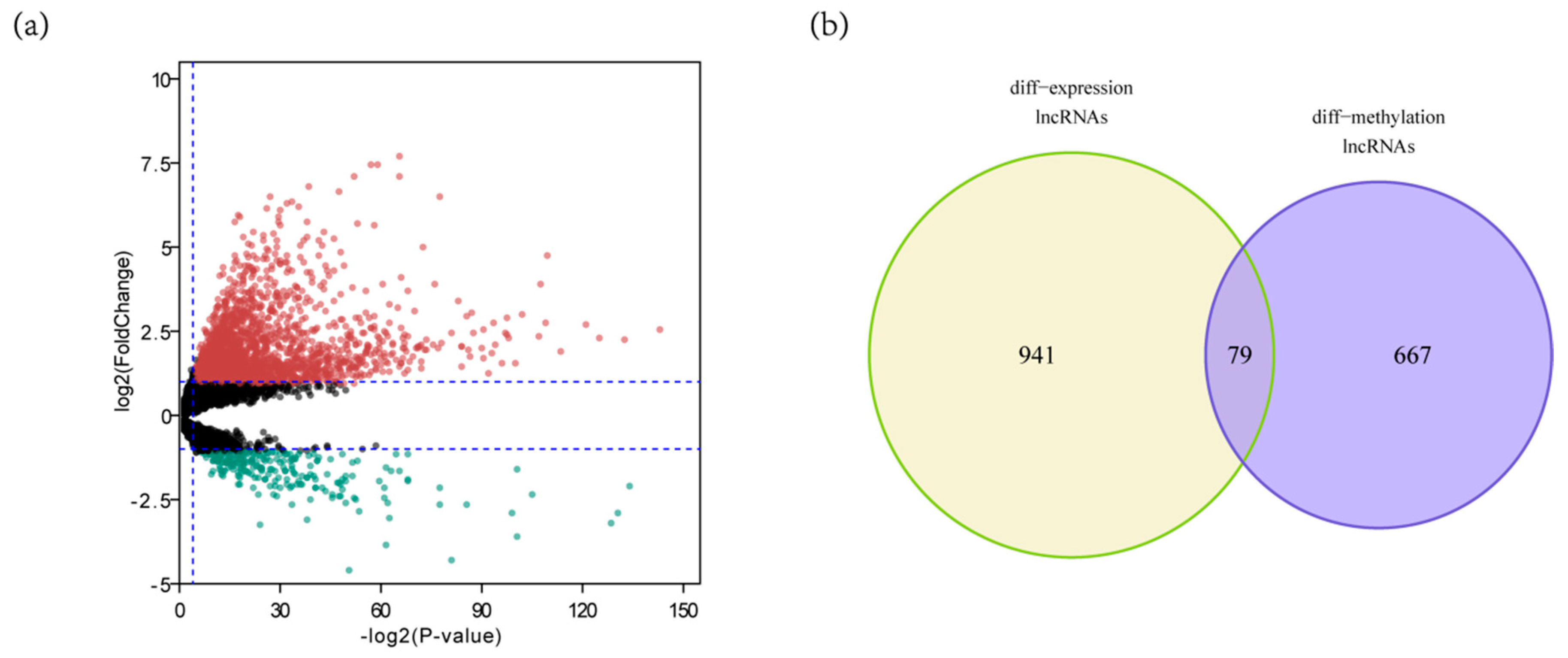
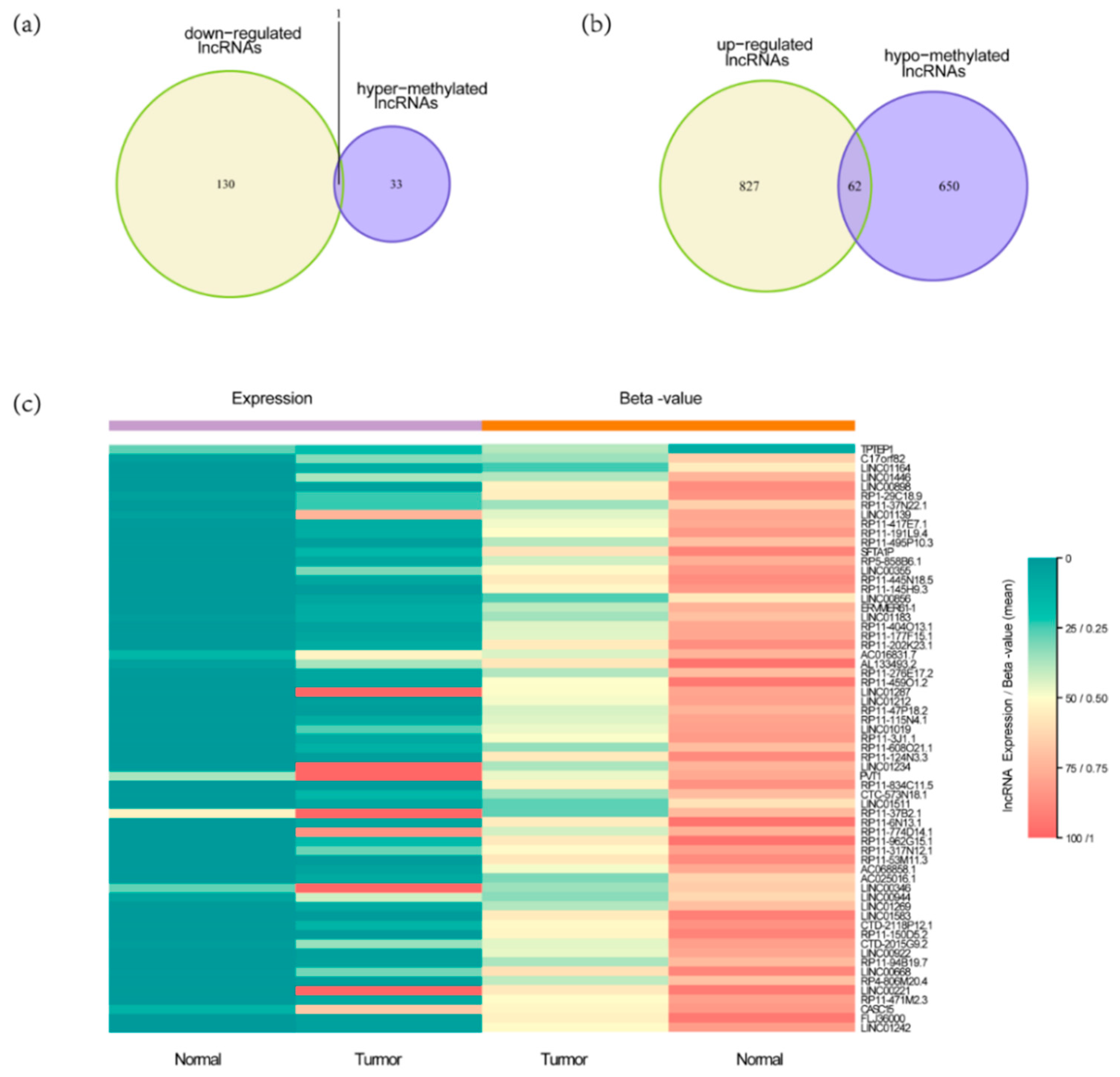
3.1. Identification of DELs with Altered lncRNAs Methylation Status in HCC
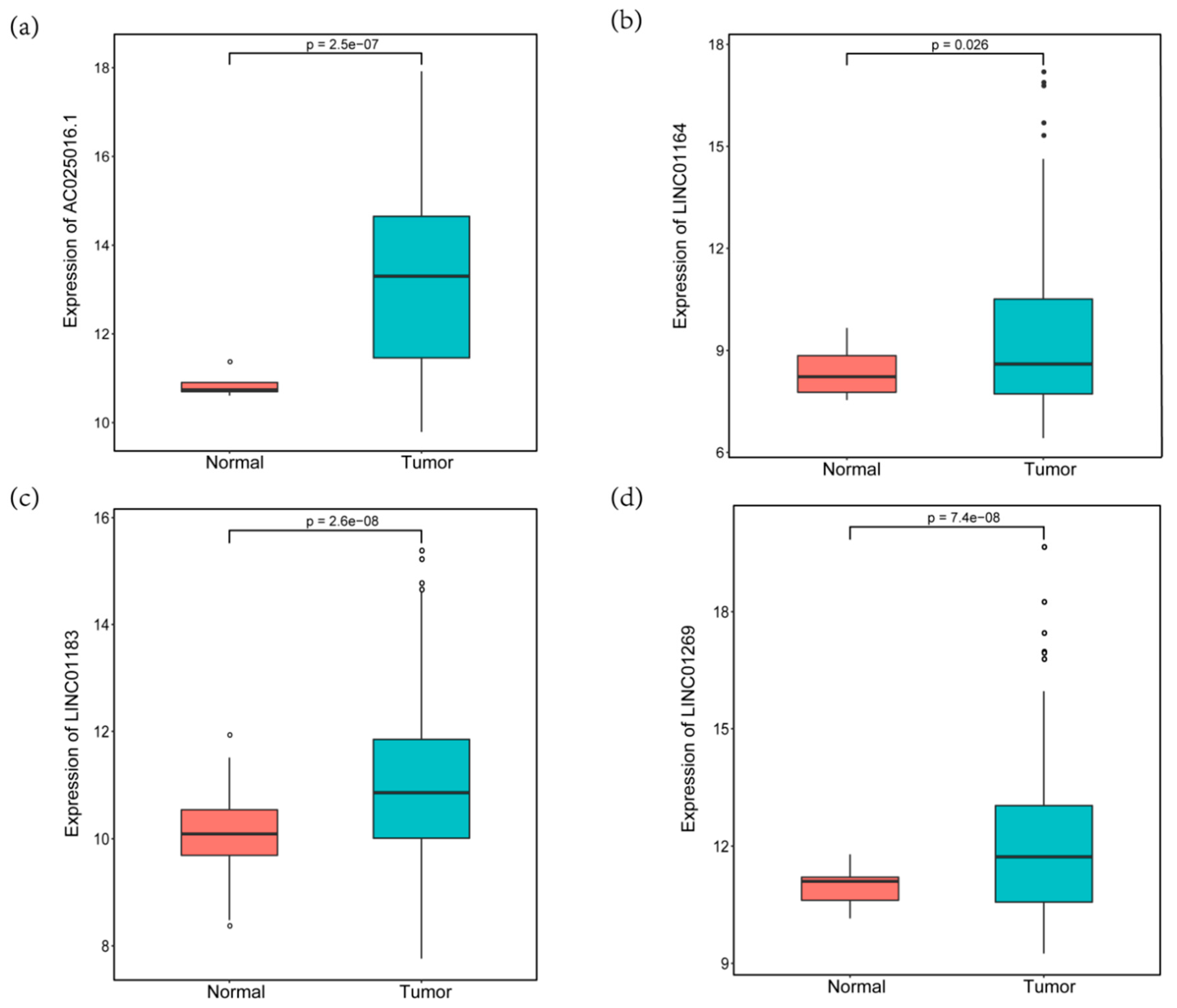
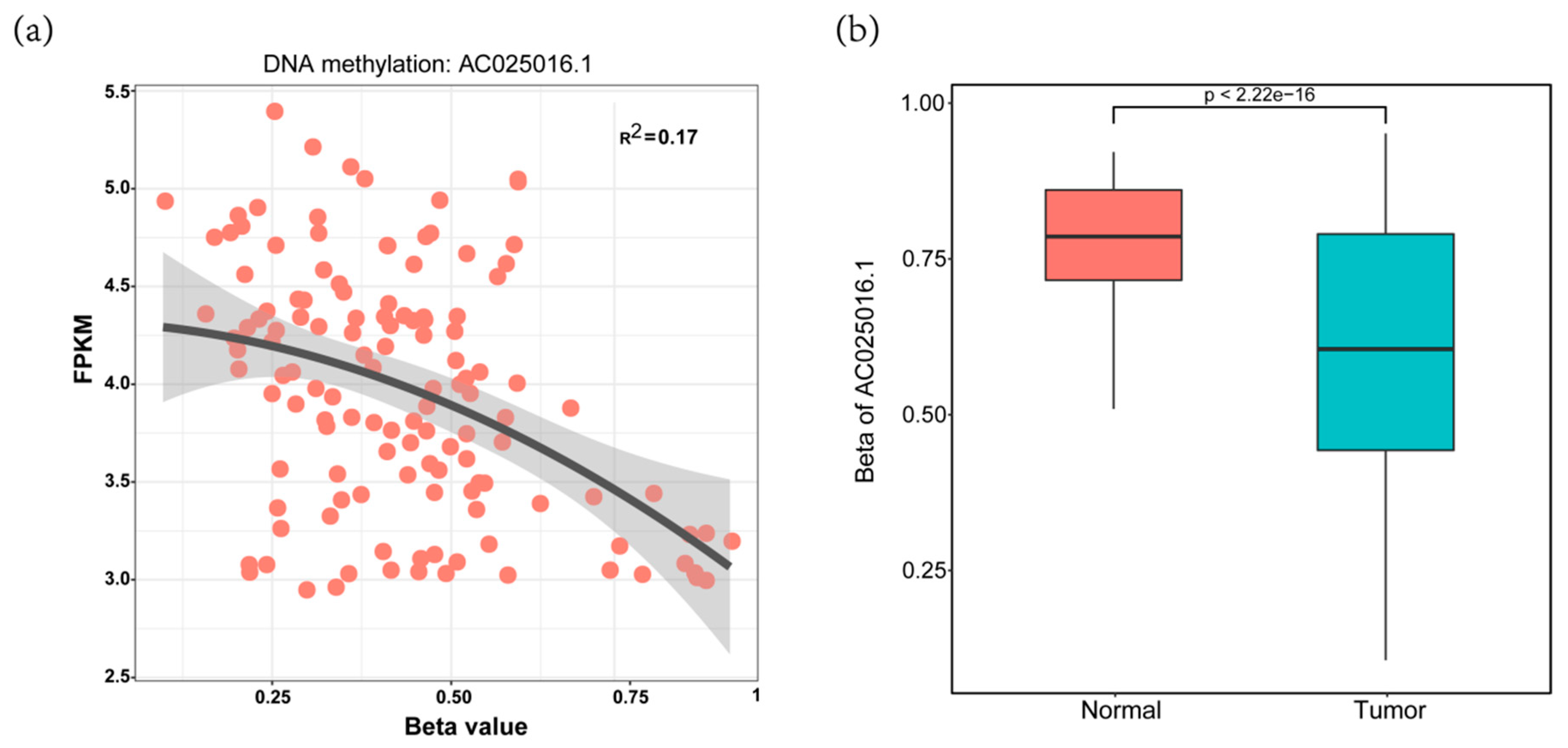
3.2. Construction of Four-MDELs HCC Prognostic Risk Score Model
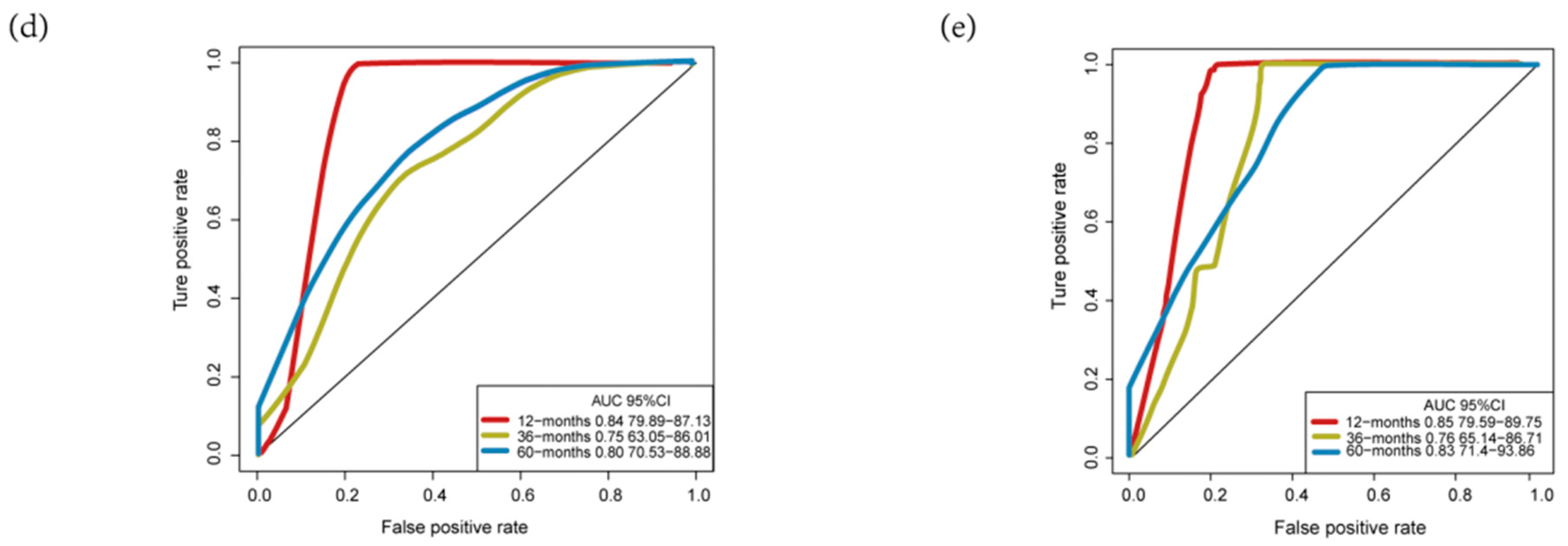
3.3. Validation and Assessment of Four-MDELs HCC Signature Prognostic Risk Score Model
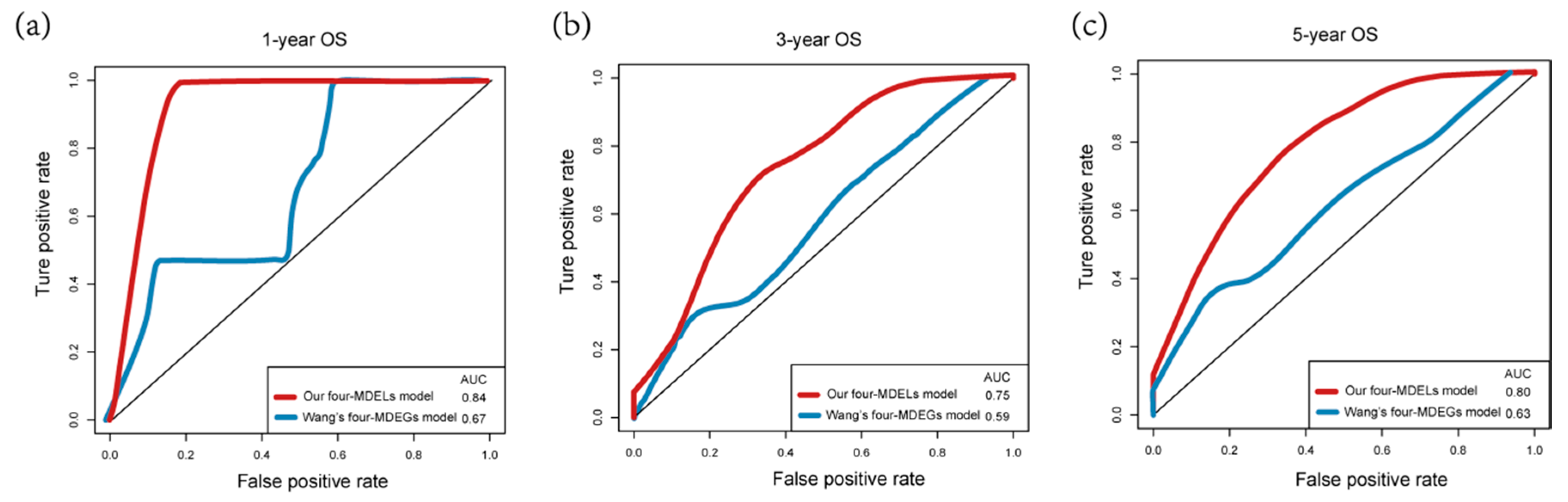
3.4. Comparisons with Other Models
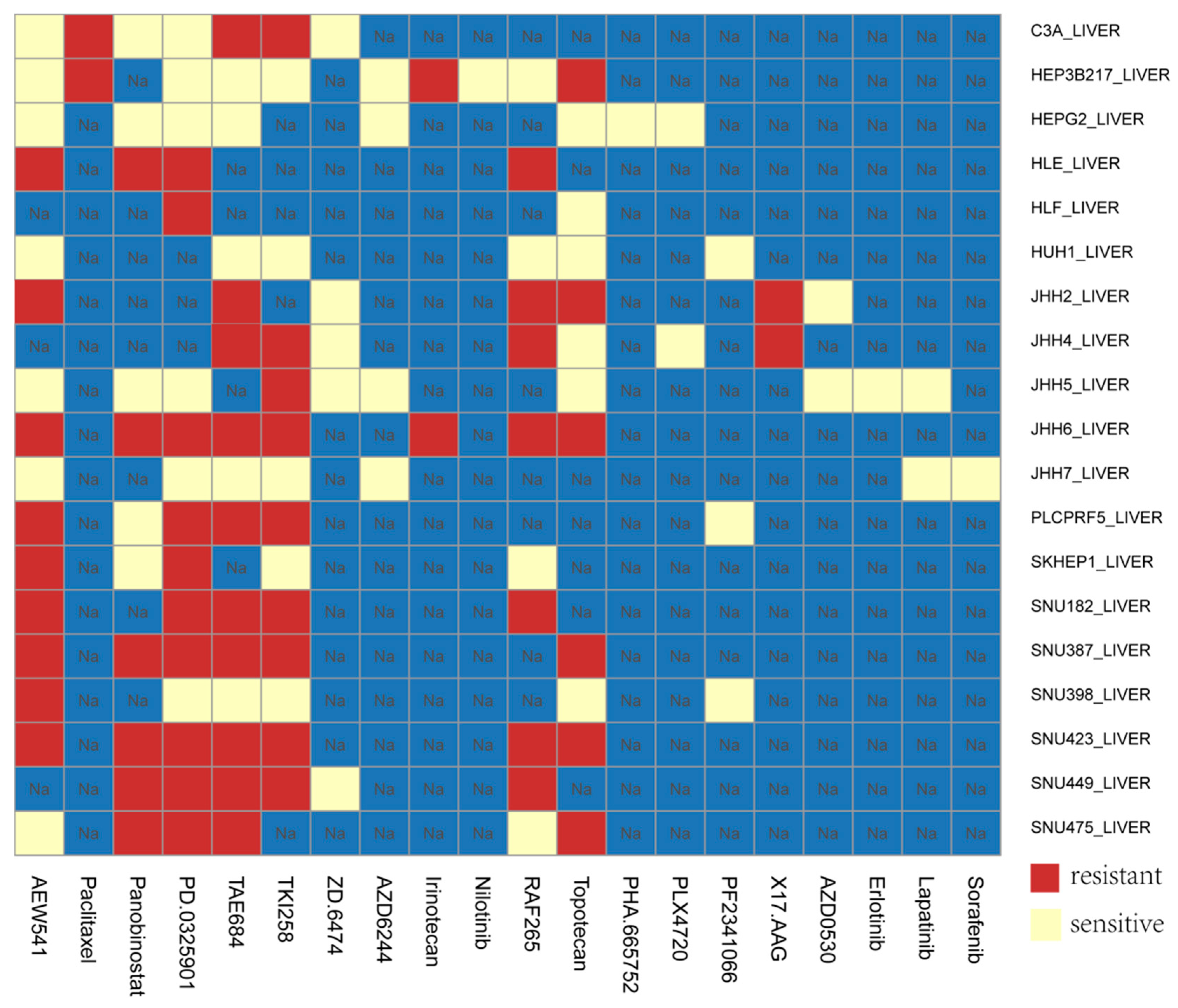
3.5. Application of Our Model in HCC Cell Lines
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Zheng, R.; Sun, K.; Zhang, S.; Zeng, H.; Zou, X.; Chen, R.; Gu, X.; Wei, W.; He, J. Report of cancer epidemiology in China, 2015. Zhonghua Zhong Liu Za Zhi 2019, 41, 19–28. [Google Scholar]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Villanueva, A.; Hernandez-Gea, V.; Llovet, J.M. Medical therapies for hepatocellular carcinoma: A critical view of the evidence. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, M.; Zhang, M.; De Wilde, R.F.; Ottenhof, N.A.; Rezaee, N.; Wolfgang, C.L.; Blackford, A.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N. Very long-term survival following resection for pancreatic cancer is not explained by commonly mutated genes: Results of whole-exome sequencing analysis. Clin. Cancer Res. 2015, 21, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Yoon, G.; Seo, A.N. What are the most important prognostic factors in patients with residual rectal cancer after preoperative chemoradiotherapy? Yeungnam Univ. J. Med. 2019, 36, 124. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Xu, W.; Waldron, J.; Siu, L.; Shen, X.; Tong, L.; Ringash, J.; Bayley, A.; Kim, J.; Hope, A. Refining American Joint Committee on Cancer Union for International Cancer Control TNM stage and prognostic groups for human papillomavirus-related oropharyngeal carcinomas. J. Clin. Oncol. 2015, 33, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, U.; Maak, M.; Schuster, T.; Künzli, B.; Langer, R.; Slotta-Huspenina, J.; Janssen, K.-P.; Friess, H.; Rosenberg, R. Prediction of prognosis is not improved by the seventh and latest edition of the TNM classification for colorectal cancer in a single-center collective. Ann. Surg. 2011, 254, 793–801. [Google Scholar] [CrossRef]
- Abrams, T.; Ben-Josef, E.; Bloomston, P.; Botha, J.; Clary, B.; Covey, A.; Curley, S.; D’Angelica, M.; Davila, R.; Ensminger, W. NCCN clinical practice guidelines in oncology: Hepatobiliary cancers. J. Natl. Compr. Cancer Netw. 2009, 7, 350–391. [Google Scholar]
- Carninci, P.; Hayashizaki, Y. Noncoding RNA transcription beyond annotated genes. Curr. Opin. Genet. Dev. 2007, 17, 139–144. [Google Scholar] [CrossRef]
- Griffiths-Jones, S. Annotating noncoding RNA genes. Annu. Rev. Genom. Hum. Genet. 2007, 8, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Rinn, J.L.; Schier, A.F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 2011, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, J.; Zhang, K.; Feng, B.; Wang, R.; Chen, L. Progress and prospects of long noncoding RNAs (lncRNAs) in hepatocellular carcinoma. Cell. Physiol. Biochem. 2015, 36, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, J.B.; Qin, Y.; Wang, W.; Wei, L.; Teng, Y.; Guo, L.; Zhang, B.; Lin, Z.; Liu, J. PROX1 promotes hepatocellular carcinoma metastasis by way of up-regulating hypoxia-inducible factor 1α expression and protein stability. Hepatology 2013, 58, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, W.; Li, T.; Yu, X.; Zhu, Y.; Ding, F.; Li, D.; Yang, T. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomed. Pharmacother. 2016, 80, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.-Q.; Li, R.-J.; Mei, J.-Z.; Li, X.-H. Down-regulation of long non-coding RNA GAS5 is associated with the prognosis of hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 4303. [Google Scholar] [PubMed]
- Borrello, M.G.; Pierotti, M.A.; Tamborini, E.; Biassoni, D.; Rizzetti, M.G.; Pilotti, S.; Della, G.P. DNA methylation of coding and non-coding regions of the human H-RAS gene in normal and tumor tissues. Oncogene 1992, 7, 269–275. [Google Scholar]
- Li, D.; Da, L.; Tang, H.; Li, T.; Zhao, M. CpG methylation plays a vital role in determining tissue-and cell-specific expression of the human cell-death-inducing DFF45-like effector A gene through the regulation of Sp1/Sp3 binding. Nucleic Acids Res. 2008, 36, 330–341. [Google Scholar] [CrossRef]
- Zhi, H.; Ning, S.; Li, X.; Li, Y.; Wu, W.; Li, X. A novel reannotation strategy for dissecting DNA methylation patterns of human long intergenic non-coding RNAs in cancers. Nucleic Acids Res. 2014, 42, 8258–8270. [Google Scholar] [CrossRef]
- Ma, X.; Yu, L.; Wang, P.; Yang, X. Discovering DNA methylation patterns for long non-coding RNAs associated with cancer subtypes. Comput. Biol. Chem. 2017, 69, 164–170. [Google Scholar] [CrossRef]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; Pontes, L.L.D.F.; Alberich-Jorda, M.; Zhang, P. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Lou, J.; Yang, Y.; Feng, J.; Hao, Y.; Huang, S.; Yin, L.; Xu, J.; Huang, D.; Ma, B. Dysfunction of the WT1-MEG3 signaling promotes AML leukemogenesis via p53-dependent and-independent pathways. Leukemia 2017, 31, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, S.; Yang, Y.; Yuan, S.-X.; Wu, F.-Q.; Wang, L.-L.; Yan, H.-L.; Sun, S.-H.; Zhou, W.-P. Epigenetically silenced long noncoding-SRHC promotes proliferation of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Braconi, C.; Kogure, T.; Valeri, N.; Huang, N.; Nuovo, G.; Costinean, S.; Negrini, M.; Miotto, E.; Croce, C.M.; Patel, T. MicroRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene 2011, 30, 4750. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Q.; Feng, S.; Zhao, Y.; Tao, C. Identification of four prognostic LncRNAs for survival prediction of patients with hepatocellular carcinoma. Peer J. 2017, 5, e3575. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, L.; Gu, J.; Zhang, H.; Yuan, J.; Lian, Q.; Lv, G.; Wang, S.; Wu, Y.; Yang, Y.-C.T. Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nat. Commun. 2017, 8, 14421. [Google Scholar] [CrossRef]
- Sproul, D.; Nestor, C.; Culley, J.; Dickson, J.H.; Dixon, J.M.; Harrison, D.J.; Meehan, R.R.; Sims, A.H.; Ramsahoye, B.H. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4364–4369. [Google Scholar] [CrossRef]
- Sproul, D.; Kitchen, R.R.; Nestor, C.E.; Dixon, J.M.; Sims, A.H.; Harrison, D.J.; Ramsahoye, B.H.; Meehan, R.R. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol. 2012, 13, 1–16. [Google Scholar] [CrossRef]
- Wang, Y.; Ruan, Z.; Yu, S.; Tian, T.; Liang, X.; Jing, L.; Li, W.; Wang, X.; Xiang, L.; Claret, F.-X. A four-methylated mRNA signature-based risk score system predicts survival in patients with hepatocellular carcinoma. Aging 2019, 11, 160. [Google Scholar] [CrossRef]
- Karaman, B.; Battal, B.; Sari, S.; Verim, S.J.W. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 18059. [Google Scholar] [CrossRef]
- Xiang, J.-F.; Yin, Q.-F.; Chen, T.; Zhang, Y.; Zhang, X.-O.; Wu, Z.; Zhang, S.; Wang, H.-B.; Ge, J.; Lu, X. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Xu, Y.; Yang, X.; Wang, J.; Hu, J.; Xu, L.; Yin, R. CCAT2 is a lung adenocarcinoma-specific long non-coding RNA and promotes invasion of non-small cell lung cancer. Tumor Biol. 2014, 35, 5375–5380. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Guo, M.; Herman, J.G.; Clark, D.P. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am. J. Pathol. 2003, 163, 1101–1107. [Google Scholar] [CrossRef]
- Hlady, R.A.; Sathyanarayan, A.; Thompson, J.J.; Zhou, D.; Wu, Q.; Pham, K.; Lee, J.H.; Liu, C.; Robertson, K.D. Integrating the Epigenome to Identify Drivers of Hepatocellular Carcinoma. Hepatology 2019, 69, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, D.; Xing, R.; Nie, H.; Mou, Y.; Zhang, Y.; Zhou, X. Identification of long noncoding RNAs biomarkers in patients with hepatitis B virus-associated hepatocellular carcinoma. Cancer Biomark. 2018, 23, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Q.; Wang, F.; Zhang, X.; Tang, Y.; Wang, S. LncRNA LINC01446 promotes glioblastoma progression by modulating miR-489-3p/TPT1 axis. Biochem. Biophys. Res. Commun. 2018, 503, 1484–1490. [Google Scholar] [CrossRef]
- Zhao, X.; Cui, L. A robust six-miRNA prognostic signature for head and neck squamous cell carcinoma. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef]
- Shen, Y.; Peng, X.; Shen, C. Identification and validation of immune-related lncRNA prognostic signature for breast cancer. Genomics 2020, 112, 2640–2646. [Google Scholar] [CrossRef]
- Chiang, J.W.; Karlan, B.Y.; Cass, L.; Baldwin, R.L. BRCA1 promoter methylation predicts adverse ovarian cancer prognosis. Gynecol. Oncol. 2006, 101, 403–410. [Google Scholar] [CrossRef]
- Shi, X.; Tan, H.; Le, X.; Xian, H.; Li, X.; Huang, K.; Luo, V.Y.; Liu, Y.; Wu, Z.; Mo, H. An expression signature model to predict lung adenocarcinoma-specific survival. Cancer Manag. Res. 2018, 10, 3717. [Google Scholar] [CrossRef]
- Huang, G.-W.; Xue, Y.-J.; Wu, Z.-Y.; Xu, X.-E.; Wu, J.-Y.; Cao, H.-H.; Zhu, Y.; He, J.-Z.; Li, C.-Q.; Li, E.-M. A three-lncRNA signature predicts overall survival and disease-free survival in patients with esophageal squamous cell carcinoma. BMC Cancer 2018, 18, 147. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Xiao, R.; Zhang, C.; Suyila, Q.; Li, X.; Su, X. Selecting lncRNAs in gastric cancer cells for directed therapy with bioactive peptides and chemotherapy drugs. Oncotarget 2017, 8, 86082. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2018, 47, D1034–D1037. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chr | Start | End |
|---|---|---|---|
| AC025016.1 | chr11 | 5,944,985 | 5,949,571 |
| LINC01183 | chr5 | 127,802,442 | 127,802,442 |
| LINC01269 | chr14 | 70,707,575 | 70,707,575 |
| LINC01164 | chr10 | 131,773,701 | 131,773,701 |
| OS | AUC (Our Model) | AUC (Wang’s Model) |
|---|---|---|
| 1-year | 0.84 | 0.67 |
| 3-year | 0.75 | 0.59 |
| 5-year | 0.80 | 0.63 |
| Characteristics | Patients | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|
| HR 1 (95% CI) 2 | p–Value | HR (95% CI) | p–Value | ||
| Gender (male/female) | 255/122 | 0.758 (0.498–1.154) | 0.196 | 0.763 (0.482–1.208) | 0.249 |
| Age (≤ 65 vs. > 65) | 235/141 | 0.666 (0.441––1.007) | 0.054 | 0.729 (0.463–1.145) | 0.17 |
| Tumor (T3–T4/T1–T2) | 94/280 | 1.794 (1.146–2.808) | 0.011 | 0.715 (0.086–5.919) | 0.756 |
| Pathologic stage (III–IV/I–II) | 91/262 | 1.483 (0.906–2.427) | 0.117 | 0.744 (0.298–1.856) | 0.526 |
| BMI 3 (≥ 25 vs. <25) | 161/180 | 0.906 (0.577–1.423) | 0.669 | ||
| Adjacent_hepatic_tissue_inflammation (no/yes) | 119/120 | 1.128 (0.662–1.923) | 0.657 | ||
| Virus 4 (no/yes) | 211/166 | 0.649 (0.428–0.985) | 0.042 | ||
| Fibrosis (no/yes) | 76/142 | 0.813 (0.474–1.397) | 0.454 | ||
| Radiation therapy (no/yes) | 243/4 | 2.127 (0.29–15.622) | 0.458 | ||
| Relative_family_cancer_history (no/yes) | 212/114 | 1.795 (1.16–2.779) | 0.009 | ||
| Pharmaceutical_therapy (no/yes) | 103/26 | 2.189 (0.951–5.04) | 0.066 | ||
| New tumor event after initial treatment (no/yes) | 149/142 | 1.592 (0.877–2.89) | 0.126 | 0.654 (0.179–2.382) | 0.519 |
| Person neoplasm cancer status (with tumor/tumor free) | 123/157 | 2.208 (1.224–3.983) | 0.009 | 2.816 (0.774–10.245) | 0.116 |
| PI (High–risk/Low–risk) | 174/174 | 1.838 (1.177–2.87) | 0.007 | 2.469 (1.238–4.923) | 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, L.-E.; Hu, D.-D.; Zheng, Y. A Four-Methylated lncRNAs-Based Prognostic Signature for Hepatocellular Carcinoma. Genes 2020, 11, 908. https://doi.org/10.3390/genes11080908
Liao L-E, Hu D-D, Zheng Y. A Four-Methylated lncRNAs-Based Prognostic Signature for Hepatocellular Carcinoma. Genes. 2020; 11(8):908. https://doi.org/10.3390/genes11080908
Chicago/Turabian StyleLiao, Le-En, Dan-Dan Hu, and Yun Zheng. 2020. "A Four-Methylated lncRNAs-Based Prognostic Signature for Hepatocellular Carcinoma" Genes 11, no. 8: 908. https://doi.org/10.3390/genes11080908
APA StyleLiao, L.-E., Hu, D.-D., & Zheng, Y. (2020). A Four-Methylated lncRNAs-Based Prognostic Signature for Hepatocellular Carcinoma. Genes, 11(8), 908. https://doi.org/10.3390/genes11080908