The Evolution of Gene Therapy in the Treatment of Metabolic Liver Diseases



Abstract
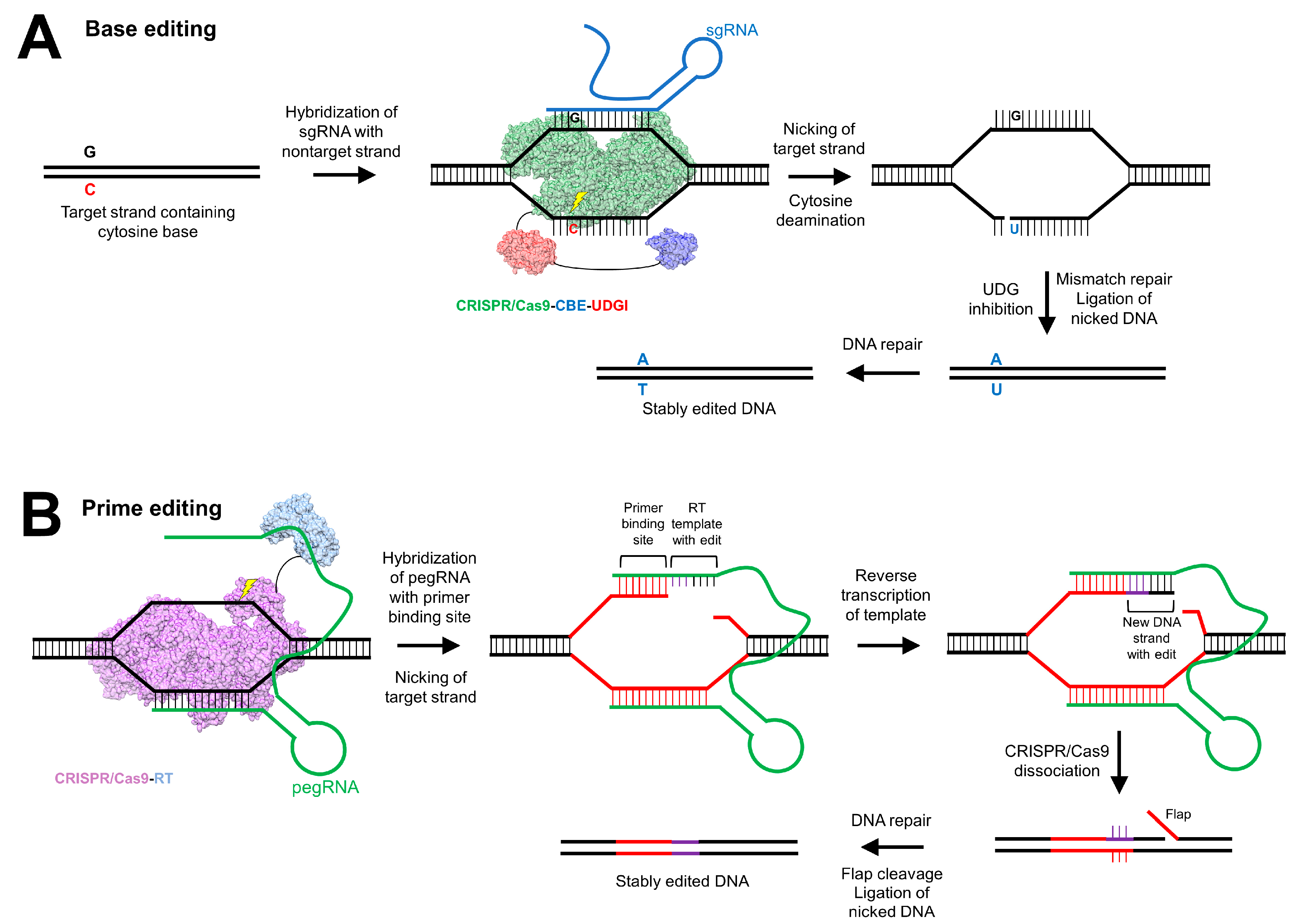
:1. Introduction
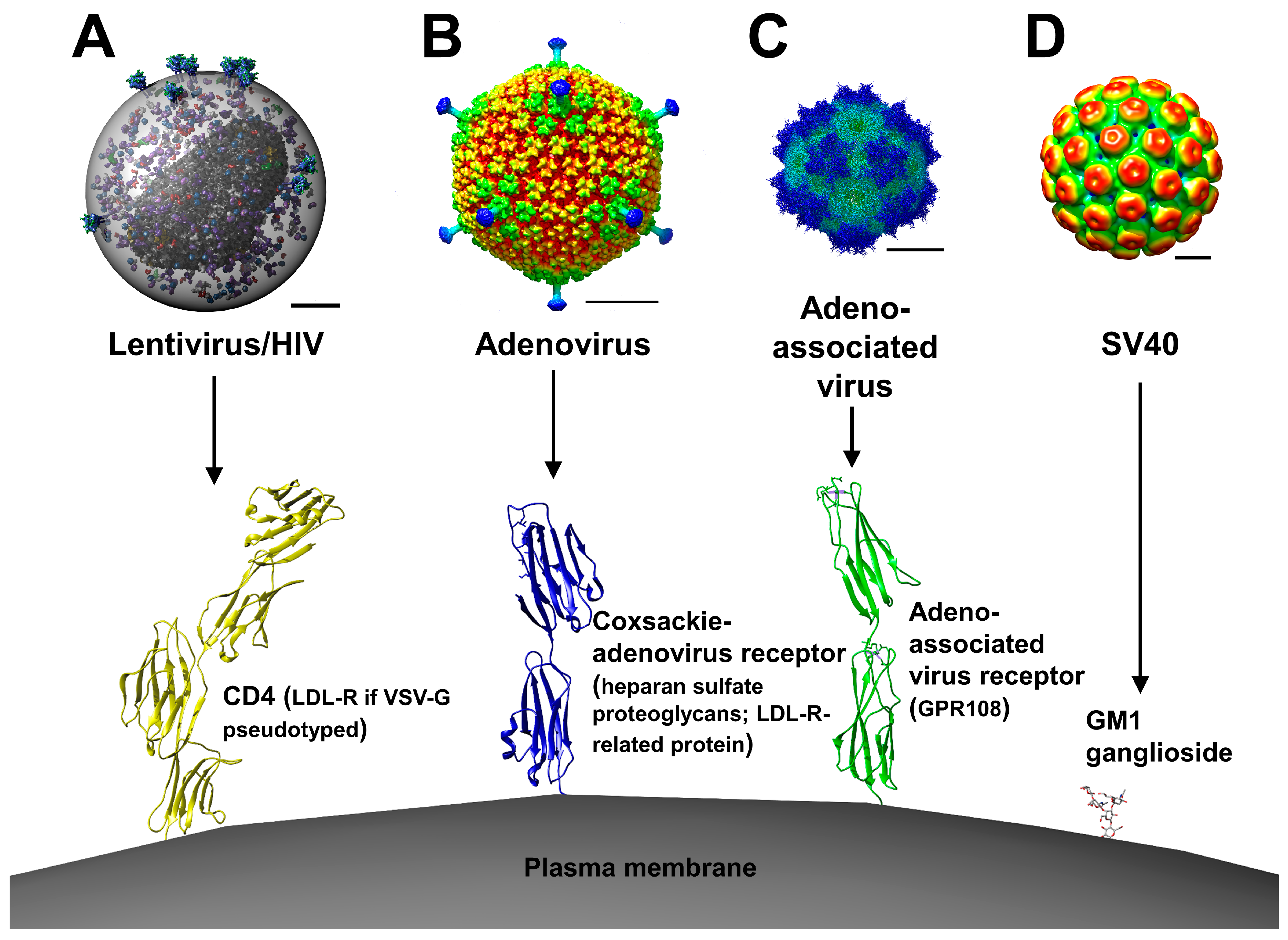
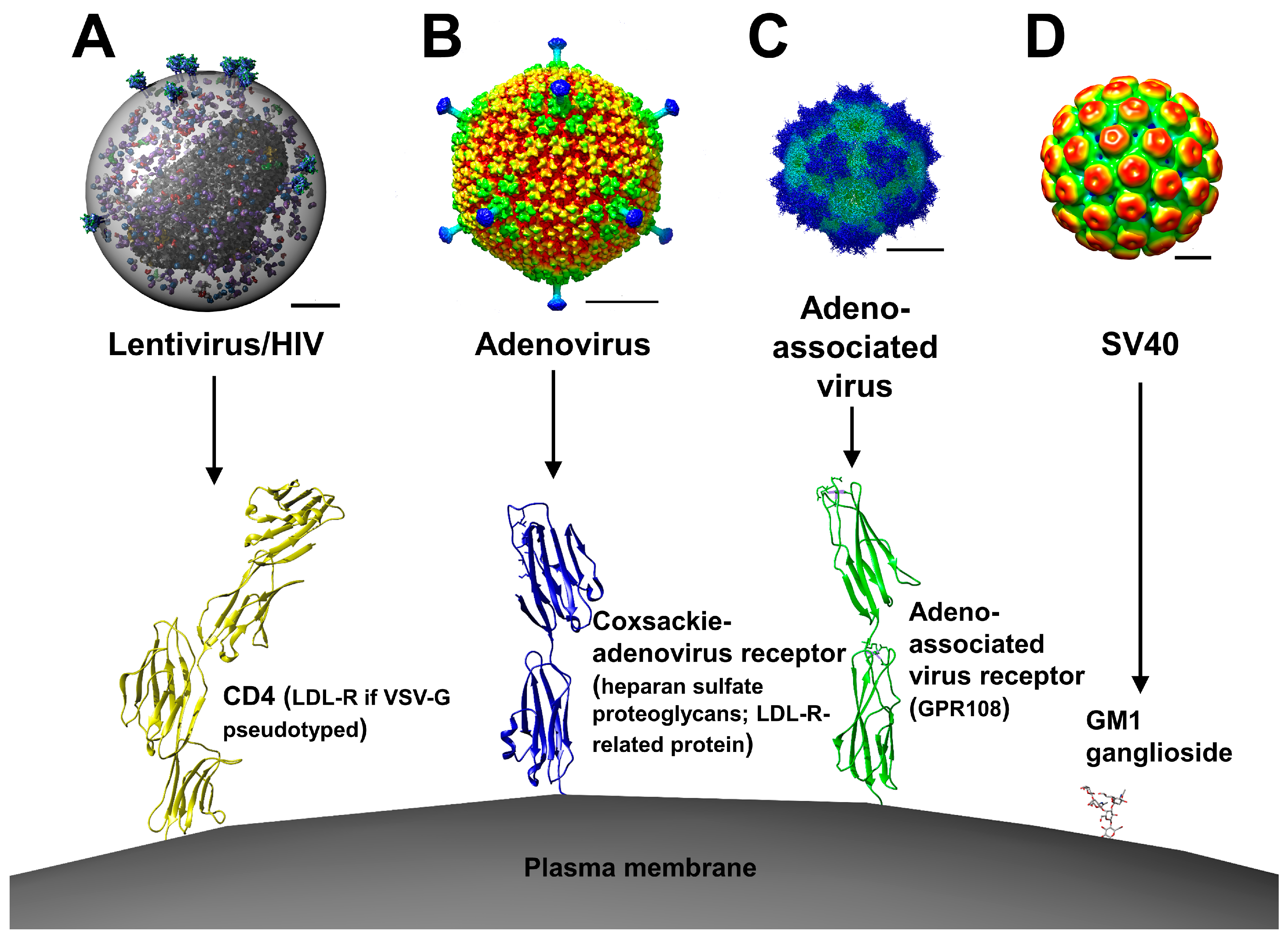
2. Viral Vectors
2.1. Retroviral and Lentiviral Vectors
2.2. Adenoviral Vectors
2.3. Adeno-Associated Viral Vectors
2.4. Simian Virus 40
3. Non-Viral Vectors
3.1. Sleeping Beauty Transposon
3.2. piggyBac Transposon
3.3. Naked Nucleic Acids and Synthetic Delivery Vectors
4. RNA-Based Therapeutics as Gene Suppression Tools
4.1. Small Interfering RNAs
4.2. microRNAs
5. Genome Editing
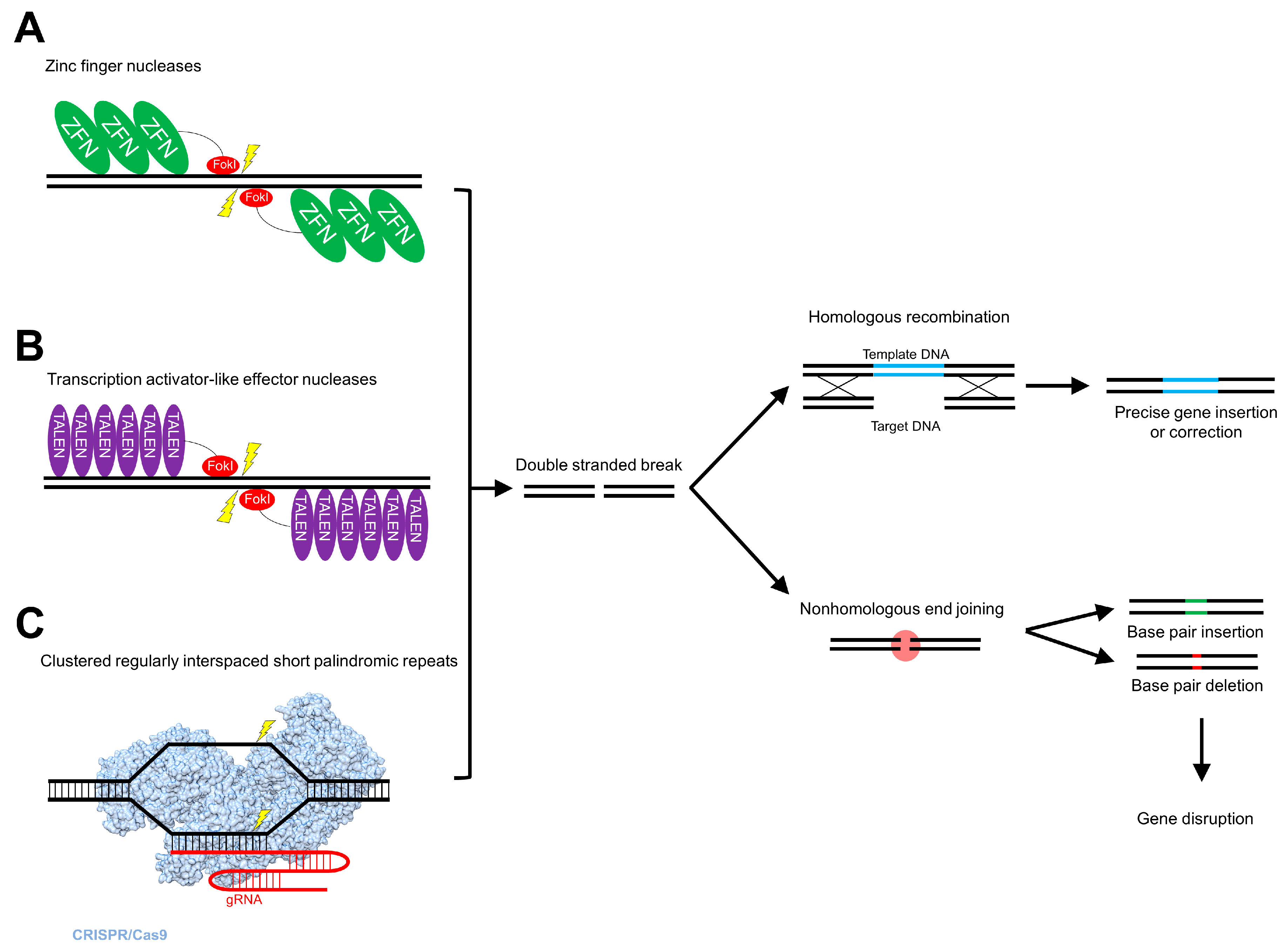
5.1. Zinc Finger Nucleases
5.2. Transcription Activator-Like Effector Nucleases
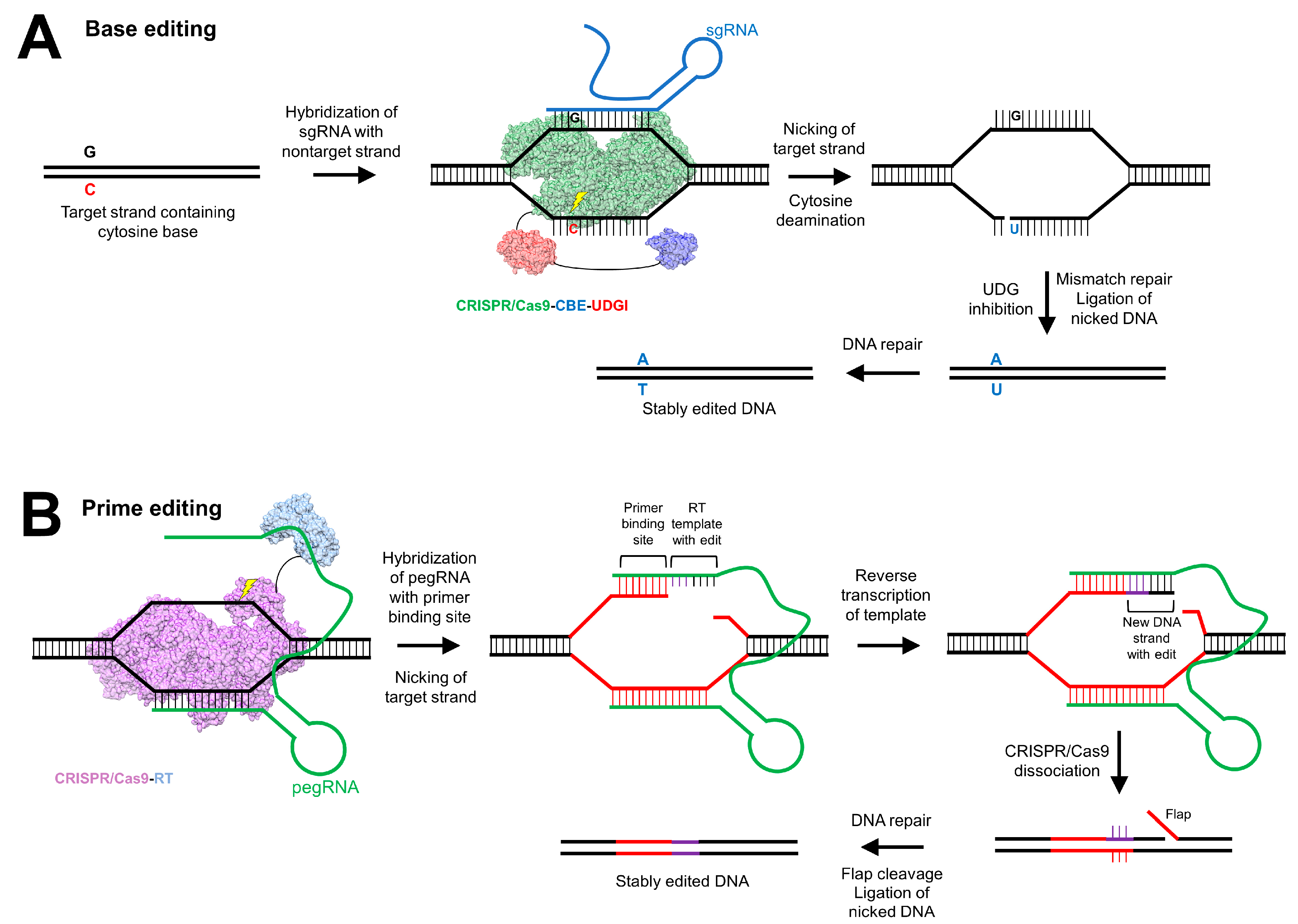
5.3. Clustered Regularly Interspaced Short Palindromic Repeats
6. Future Perspectives
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AAT | α-1 antitrypsin |
| AAV | adeno-associated virus |
| AAVR | adeno-associated virus receptor |
| AGXT | alanine-glyoxylate aminotransferase |
| Arg1 | arginase-1 |
| Cas | CRISPR-associated gene |
| cccDNA | covalently closed circular DNA |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| DNA | deoxyribonucleic acid |
| DSB | double-stranded break |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| HDAd | helper-dependent adenovirus |
| HIV | human immunodeficiency virus |
| HR | homologous recombination |
| iHLC | induced hepatocyte-like cell |
| LTGT | liver-targeted gene therapy |
| MLV | murine leukemia virus |
| M-MLV | Moloney murine leukemia virus |
| NASH | non-alcoholic steatohepatitis |
| NHEJ | nonhomologous end joining |
| pegRNA | prime editing guide RNA |
| PH1 | primary hyperoxaluria type 1 |
| rAAV | recombinant adeno-associated virus |
| rAd | recombinant adenovirus |
| RNA | ribonucleic acid |
| rSV40 | recombinant simian virus 40 |
| SB | Sleeping Beauty |
| Sp | Streptococcus pyogenes |
| St | Streptococcus thermophilus |
| SV40 | simian virus 40 |
| TALEN | transcription activator-like effector nuclease |
| Tag | T cell antigen |
| TERT | telomerase reverse transcriptase |
| UGT1A1 | uridine diphosphate glucuronosyltransferase family 1 member A1 |
| ZFN | zinc-finger nuclease |
References
- Qi, P.; Han, J.; Lu, Y.; Wang, C.; Zhu, B. A transient three-plasmid expression system for the production of hepatocytes targeting retroviral vectors. Acta Biochim. Biophys. Sin. 2007, 39, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Loux, N.; Dagher, I.; Vons, C.; Carey, K.; Briand, P.; Hadchouel, M.; Franco, D.; Jouanneau, J.; Schwall, R.; et al. Improved gene transfer selectivity to hepatocarcinoma cells by retrovirus vector displaying single-chain variable fragment antibody against c-Met. Cancer Gene Ther. 2003, 10, 840–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Pagès, J.C.; Farge, D.; Briand, P.; Weber, A. Amphotropic retroviral vectors displaying hepatocyte growth factor-envelope fusion proteins improve transduction efficiency of primary hepatocytes. Hum. Gene Ther. 1998, 9, 2469–2479. [Google Scholar] [CrossRef]
- Liu, M.L.; Winther, B.L.; Kay, M.A. Pseudotransduction of hepatocytes by using concentrated pseudotyped vesicular stomatitis virus G glycoprotein (VSV-G)-Moloney murine leukemia virus-derived retrovirus vectors: Comparison of VSV-G and amphotropic vectors for hepatic gene transfer. J. Virol. 1996, 70, 2497–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.G.; Adam, M.A.; Miller, A.D. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell. Biol. 1990, 10, 4239–4242. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Mei, M.; Ma, X.; Ponder, K.P. High expression reduces an antibody response after neonatal gene therapy with B domain-deleted human factor VIII in mice. J. Thromb. Haemost. 2007, 5, 1805–1812. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Young, G.R.; Stoye, J.P.; Kassiotis, G. Are human endogenous retroviruses pathogenic? An approach to testing the hypothesis. Bioessays 2013, 35, 794–803. [Google Scholar] [CrossRef]
- Andreadis, S.; Palsson, B.O. Coupled effects of polybrene and calf serum on the efficiency of retroviral transduction and the stability of retroviral vectors. Hum. Gene Ther. 1997, 8, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Ryser, M.F.; Roesler, J.; Gentsch, M.; Brenner, S. Gene therapy for chronic granulomatous disease. Expert Opin. Biol. Ther. 2007, 7, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Le Doux, J.M.; Davis, H.E.; Morgan, J.R.; Yarmush, M.L. Kinetics of retrovirus production and decay. Biotechnol. Bioeng. 1999, 63, 654–662. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.S.; Schiedlmeier, B.; Brugman, M.H.; Stahlhut, M.; Bartels, S.; Li, Z.; Baum, C. Cell-intrinsic and vector-related properties cooperate to determine the incidence and consequences of insertional mutagenesis. Mol. Ther. 2009, 17, 1537–1547. [Google Scholar] [CrossRef] [Green Version]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knöss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [Green Version]
- Kafri, T.; van Praag, H.; Ouyang, L.; Gage, F.H.; Verma, I.M. A packaging cell line for lentivirus vectors. J. Virol. 1999, 73, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Akkina, R.K.; Walton, R.M.; Chen, M.L.; Li, Q.X.; Planelles, V.; Chen, I.S. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J. Virol. 1996, 70, 2581–2585. [Google Scholar] [CrossRef] [Green Version]
- DePolo, N.J.; Reed, J.D.; Sheridan, P.L.; Townsend, K.; Sauter, S.L.; Jolly, D.J.; Dubensky, T.W. VSV-G pseudotyped lentiviral vector particles produced in human cells are inactivated by human serum. Mol. Ther. 2000, 2, 218–222. [Google Scholar] [CrossRef]
- Hu, S.; Mohan Kumar, D.; Sax, C.; Schuler, C.; Akkina, R. Pseudotyping of lentiviral vector with novel vesiculovirus envelope glycoproteins derived from Chandipura and Piry viruses. Virology 2016, 488, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Milani, M.; Annoni, A.; Moalli, F.; Liu, T.; Cesana, D.; Calabria, A.; Bartolaccini, S.; Biffi, M.; Russo, F.; Visigalli, I.; et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Mátrai, J.; Cantore, A.; Bartholomae, C.C.; Annoni, A.; Wang, W.; Acosta-Sanchez, A.; Samara-Kuko, E.; De Waele, L.; Ma, L.; Genovese, P.; et al. Hepatocyte-targeted expression by integrase-defective lentiviral vectors induces antigen-specific tolerance in mice with low genotoxic risk. Hepatology 2011, 53, 1696–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.M.; Bassit, L.C.; Mueller, H.; Kornepati, A.V.R.; Bogerd, H.P.; Nie, T.; Chatterjee, P.; Javanbakht, H.; Schinazi, R.F.; Cullen, B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 2015, 476, 196–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, A.; Kay, M.A. A new adenoviral helper-dependent vector results in long-term therapeutic levels of human coagulation factor IX at low doses in vivo. Blood 2002, 99, 3923–3930. [Google Scholar] [CrossRef]
- Ehrhardt, A.; Xu, H.; Kay, M.A. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 2003, 77, 7689–7695. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, F.; Pastore, N.; Abarrategui-Pontes, C.; Flageul, M.; Myara, A.; Laplanche, S.; Labrune, P.; Podevin, G.; Nguyen, T.H.; Brunetti-Pierri, N. Correction of hyperbilirubinemia in Gunn rats by surgical delivery of low doses of helper-dependent adenoviral vectors. Hum. Gene Ther. Methods 2014, 25, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Marrone, J.; Lehmann, G.L.; Soria, L.R.; Pellegrino, J.M.; Molinas, S.; Marinelli, R.A. Adenoviral transfer of human aquaporin-1 gene to rat liver improves bile flow in estrogen-induced cholestasis. Gene Ther. 2014, 21, 1058–1064. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Jiang, L.; Shan, A.; Su, Y.; Cheng, Y.; Song, D.; Ji, H.; Ning, G.; Wang, W.; Cao, Y. Targeting hepatic miR-221/222 for therapeutic intervention of nonalcoholic steatohepatitis in mice. EBioMedicine 2018, 37, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Casimiro, D.R.; Chen, L.; Fu, T.M.; Evans, R.K.; Caulfield, M.J.; Davies, M.E.; Tang, A.; Chen, M.; Huang, L.; Harris, V.; et al. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J. Virol. 2003, 77, 6305–6313. [Google Scholar] [CrossRef] [Green Version]
- Casimiro, D.R.; Bett, A.J.; Fu, T.M.; Davies, M.E.; Tang, A.; Wilson, K.A.; Chen, M.; Long, R.; McKelvey, T.; Chastain, M.; et al. Heterologous human immunodeficiency virus type 1 priming-boosting immunization strategies involving replication-defective adenovirus and poxvirus vaccine vectors. J. Virol. 2004, 78, 11434–11438. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, Q.; Ertl, H.C.; Wilson, J.M. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J. Virol. 1995, 69, 2004–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morral, N.; O’Neal, W.; Rice, K.; Leland, M.; Kaplan, J.; Piedra, P.A.; Zhou, H.; Parks, R.J.; Velji, R.; Aguilar-Córdova, E.; et al. Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc. Natl. Acad. Sci. USA 1999, 96, 12816–12821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, K.; Graham, F.L.; Caskey, C.T.; Kochanek, S. Rescue, propagation, and partial purification of a helper virus-dependent adenovirus vector. Proc. Natl. Acad. Sci. USA 1995, 92, 3854–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morral, N.; Parks, R.J.; Zhou, H.; Langston, C.; Schiedner, G.; Quinones, J.; Graham, F.L.; Kochanek, S.; Beaudet, A.L. High doses of a helper-dependent adenoviral vector yield supraphysiological levels of alpha1-antitrypsin with negligible toxicity. Hum. Gene Ther. 1998, 9, 2709–2716. [Google Scholar] [CrossRef]
- Harui, A.; Suzuki, S.; Kochanek, S.; Mitani, K. Frequency and stability of chromosomal integration of adenovirus vectors. J. Virol. 1999, 73, 6141–6146. [Google Scholar] [CrossRef] [Green Version]
- Ross, P.J.; Kennedy, M.A.; Parks, R.J. Host cell detection of noncoding stuffer DNA contained in helper-dependent adenovirus vectors leads to epigenetic repression of transgene expression. J. Virol. 2009, 83, 8409–8417. [Google Scholar] [CrossRef] [Green Version]
- Alba, R.; Bosch, A.; Chillon, M. Gutless adenovirus: Last-generation adenovirus for gene therapy. Gene Ther. 2005, 12 (Suppl. 1), S18–S27. [Google Scholar] [CrossRef] [Green Version]
- Ledgerwood, J.E.; DeZure, A.D.; Stanley, D.A.; Coates, E.E.; Novik, L.; Enama, M.E.; Berkowitz, N.M.; Hu, Z.; Joshi, G.; Ploquin, A.; et al. Chimpanzee adenovirus vector Ebola vaccine. N. Engl. J. Med. 2017, 376, 928–938. [Google Scholar] [CrossRef]
- Hausl, M.A.; Zhang, W.; Müther, N.; Rauschhuber, C.; Franck, H.G.; Merricks, E.P.; Nichols, T.C.; Kay, M.A.; Ehrhardt, A. Hyperactive Sleeping Beauty transposase enables persistent phenotypic correction in mice and a canine model for hemophilia B. Mol. Ther. 2010, 18, 1896–1906. [Google Scholar] [CrossRef]
- Hausl, M.; Zhang, W.; Voigtländer, R.; Müther, N.; Rauschhuber, C.; Ehrhardt, A. Development of adenovirus hybrid vectors for Sleeping Beauty transposition in large mammals. Curr. Gene Ther. 2011, 11, 363–374. [Google Scholar] [CrossRef]
- Castello, R.; Borzone, R.; D’Aria, S.; Annunziata, P.; Piccolo, P.; Brunetti-Pierri, N. Helper-dependent adenoviral vectors for liver-directed gene therapy of primary hyperoxaluria type 1. Gene Ther. 2016, 23, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Walsh, L.; Lester, W.; Perry, D.; Madan, B.; Laffan, M.; Yu, H.; Vettermann, C.; Pierce, G.F.; Wong, W.Y.; et al. AAV5-Factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017, 377, 2519–2530. [Google Scholar] [CrossRef]
- Ellis, B.L.; Hirsch, M.L.; Barker, J.C.; Connelly, J.P.; Steininger, R.J.; Porteus, M.H. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol. J. 2013, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Wang, Y.; Zhang, Y.; Ejjigani, A.; Yin, Z.; Lu, Y.; Wang, L.; Wang, M.; Li, J.; Hu, Z.; et al. Selective in vivo targeting of human liver tumors by optimized AAV3 vectors in a murine xenograft model. Hum. Gene Ther. 2014, 25, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Nakai, H.; Yant, S.R.; Storm, T.A.; Fuess, S.; Meuse, L.; Kay, M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001, 75, 6969–6976. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Lee, J.S.; Wang, L.; Desai, T.; Akache, B.; Storm, T.A.; Kay, M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008, 82, 5887–5911. [Google Scholar] [CrossRef] [Green Version]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.I.; Byrne, B.J.; Flotte, T.R.; Gao, G.; Hauswirth, W.W.; Herzog, R.W.; Muzyczka, N.; Vanden Driessche, T.; Xiao, X.; Zolotukhin, S.; et al. Adeno-associated virus type 2 and hepatocellular carcinoma? Hum. Gene Ther. 2015, 26, 779–781. [Google Scholar] [CrossRef] [Green Version]
- Büning, H.; Schmidt, M. Adeno-associated vector toxicity-to be or not to be? Mol. Ther. 2015, 23, 1673–1675. [Google Scholar] [CrossRef] [Green Version]
- Park, K.J.; Lee, J.; Park, J.H.; Joh, J.W.; Kwon, C.H.; Kim, J.W. Adeno-associated virus 2-mediated hepatocellular carcinoma is very rare in Korean patients. Ann. Lab. Med. 2016, 36, 469–474. [Google Scholar] [CrossRef]
- La Bella, T.; Imbeaud, S.; Peneau, C.; Mami, I.; Datta, S.; Bayard, Q.; Caruso, S.; Hirsch, T.Z.; Calderaro, J.; Morcrette, G.; et al. Adeno-associated virus in the liver: Natural history and consequences in tumour development. Gut 2020, 69, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.C.; Mami, I.; La Bella, T.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; et al. Wild-type AAV insertions in hepatocellular carcinoma do not inform debate over genotoxicity risk of vectorized AAV. Mol. Ther. 2016, 24, 660–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo, O.; Luqui, D.M.; Gazquez, C.; Martinez-Espartosa, D.; Navarro-Blasco, I.; Monreal, J.I.; Guembe, L.; Moreno-Cermeño, A.; Corrales, F.J.; Prieto, J.; et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J. Hepatol. 2016, 64, 419–426. [Google Scholar] [CrossRef]
- Yasuda, M.; Bishop, D.F.; Fowkes, M.; Cheng, S.H.; Gan, L.; Desnick, R.J. AAV8-mediated gene therapy prevents induced biochemical attacks of acute intermittent porphyria and improves neuromotor function. Mol. Ther. 2010, 18, 17–22. [Google Scholar] [CrossRef]
- Unzu, C.; Sampedro, A.; Mauleón, I.; Alegre, M.; Beattie, S.G.; de Salamanca, R.E.; Snapper, J.; Twisk, J.; Petry, H.; González-Aseguinolaza, G.; et al. Sustained enzymatic correction by rAAV-mediated liver gene therapy protects against induced motor neuropathy in acute porphyria mice. Mol. Ther. 2011, 19, 243–250. [Google Scholar] [CrossRef]
- Guenzel, A.J.; Collard, R.; Kraus, J.P.; Matern, D.; Barry, M.A. Long-term sex-biased correction of circulating propionic acidemia disease markers by adeno-associated virus vectors. Hum. Gene Ther. 2015, 26, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, R.J.; Venditti, C.P. Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA). Mol. Genet. Metab. 2012, 107, 617–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, H.; Ogura, T.; Mizukami, H.; Urabe, M.; Hamada, H.; Yoshikawa, H.; Ozawa, K.; Kume, A. Complete restoration of phenylalanine oxidation in phenylketonuria mouse by a self-complementary adeno-associated virus vector. J. Gene Med. 2011, 13, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Yiu, W.H.; Lee, Y.M.; Peng, W.T.; Pan, C.J.; Mead, P.A.; Mansfield, B.C.; Chou, J.Y. Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol. Ther. 2010, 18, 1076–1084. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Correia, C.E.; Conlon, T.; Specht, A.; Verstegen, J.; Onclin-Verstegen, K.; Campbell-Thompson, M.; Dhaliwal, G.; Mirian, L.; Cossette, H.; et al. Adeno-associated virus-mediated correction of a canine model of glycogen storage disease type Ia. Hum. Gene Ther. 2010, 21, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Aronson, S.J.; Bakker, R.S.; Shi, X.; Duijst, S.; Ten Bloemendaal, L.; de Waart, D.R.; Verheij, J.; Ronzitti, G.; Oude Elferink, R.P.; Beuers, U.; et al. Liver-directed gene therapy results in long-term correction of progressive familial intrahepatic cholestasis type 3 in mice. J. Hepatol. 2019, 71, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, D.E.; Lange, A.M.; Altynova, E.S.; Sarkar, R.; Zhou, S.; Merricks, E.P.; Franck, H.G.; Nichols, T.C.; Arruda, V.R.; Kazazian, H.H. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol. Ther. 2011, 19, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.N.; Everett, J.K.; Raymond, H.; Kafle, S.; Merricks, E.P.; Kazazian, H.H.; Nichols, T.C.; Bushman, F.D.; Sabatino, D.E. Long-term AAV-mediated factor VIII expression in nine hemophilia A dogs: A 10 year follow-up analysis on durability, safety and vector integration. Blood 2019, 134, 611. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020, 382, 29–40. [Google Scholar] [CrossRef] [PubMed]
- D’Avola, D.; López-Franco, E.; Sangro, B.; Pañeda, A.; Grossios, N.; Gil-Farina, I.; Benito, A.; Twisk, J.; Paz, M.; Ruiz, J.; et al. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J. Hepatol. 2016, 65, 776–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Day, J.W.; Chiriboga, C.A.; Crawford, T.O.; Darras, B.T.; Finkel, R.S.; Connolly, A.M.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec-xioi gene-replacement therapy for spinal muscular atrophy type 1 (SMA1): Phase 3 US study (STR1VE) update (1828). Neurology 2020, 94, 1828. [Google Scholar]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Rasko, J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Arruda, V.R.; Stedman, H.H.; Haurigot, V.; Buchlis, G.; Baila, S.; Favaro, P.; Chen, Y.; Franck, H.G.; Zhou, S.; Wright, J.F.; et al. Peripheral transvenular delivery of adeno-associated viral vectors to skeletal muscle as a novel therapy for hemophilia B. Blood 2010, 115, 4678–4688. [Google Scholar] [CrossRef] [Green Version]
- Haurigot, V.; Mingozzi, F.; Buchlis, G.; Hui, D.J.; Chen, Y.; Basner-Tschakarjan, E.; Arruda, V.R.; Radu, A.; Franck, H.G.; Wright, J.F.; et al. Safety of AAV factor IX peripheral transvenular gene delivery to muscle in hemophilia B dogs. Mol. Ther. 2010, 18, 1318–1329. [Google Scholar] [CrossRef]
- Jiang, H.; Couto, L.B.; Patarroyo-White, S.; Liu, T.; Nagy, D.; Vargas, J.A.; Zhou, S.; Scallan, C.D.; Sommer, J.; Vijay, S.; et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood 2006, 108, 3321–3328. [Google Scholar] [CrossRef] [Green Version]
- Scallan, C.D.; Jiang, H.; Liu, T.; Patarroyo-White, S.; Sommer, J.M.; Zhou, S.; Couto, L.B.; Pierce, G.F. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood 2006, 107, 1810–1817. [Google Scholar] [CrossRef] [Green Version]
- Sowd, G.A.; Fanning, E. A wolf in sheep’s clothing: SV40 co-opts host genome maintenance proteins to replicate viral DNA. PLoS Pathog. 2012, 8, e1002994. [Google Scholar] [CrossRef]
- Strayer, D.; Branco, F.; Zern, M.A.; Yam, P.; Calarota, S.A.; Nichols, C.N.; Zaia, J.A.; Rossi, J.; Li, H.; Parashar, B.; et al. Durability of transgene expression and vector integration: Recombinant SV40-derived gene therapy vectors. Mol. Ther. 2002, 6, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Sauter, B.V.; Parashar, B.; Chowdhury, N.R.; Kadakol, A.; Ilan, Y.; Singh, H.; Milano, J.; Strayer, D.S.; Chowdhury, J.R. A replication-deficient rSV40 mediates liver-directed gene transfer and a long-term amelioration of jaundice in Gunn rats. Gastroenterology 2000, 119, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Vera, M.; Prieto, J.; Strayer, D.S.; Fortes, P. Factors influencing the production of recombinant SV40 vectors. Mol. Ther. 2004, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Fahrbach, K.M.; Katzman, R.B.; Rundell, K. Role of SV40 ST antigen in the persistent infection of mesothelial cells. Virology 2008, 370, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Toscano, M.G.; van der Velden, J.; van der Werf, S.; Odijk, M.; Roque, A.; Camacho-Garcia, R.J.; Herrera-Gomez, I.G.; Mancini, I.; de Haan, P. Generation of a Vero-based packaging cell line to produce SV40 gene delivery vectors for use in clinical gene therapy studies. Mol. Ther. Methods Clin. Dev. 2017, 6, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, G.T.; Autin, L.; Al-Alusi, M.; Goodsell, D.S.; Sanner, M.F.; Olson, A.J. cellPACK: A virtual mesoscope to model and visualize structural systems biology. Nat. Methods 2015, 12, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kwong, P.D.; Hendrickson, W.A. Dimeric association and segmental variability in the structure of human CD4. Nature 1997, 387, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Flatt, J.W.; Kim, R.; Smith, J.G.; Nemerow, G.R.; Stewart, P.L. An intrinsically disordered region of the adenovirus capsid is implicated in neutralization by human alpha defensin 5. PLoS ONE 2013, 8, e61571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patzke, C.; Max, K.E.; Behlke, J.; Schreiber, J.; Schmidt, H.; Dorner, A.A.; Kröger, S.; Henning, M.; Otto, A.; Heinemann, U.; et al. The coxsackievirus-adenovirus receptor reveals complex homophilic and heterophilic interactions on neural cells. J. Neurosci. 2010, 30, 2897–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.Z.; Aiyer, S.; Mietzsch, M.; Hull, J.A.; McKenna, R.; Grieger, J.; Samulski, R.J.; Baker, T.S.; Agbandje-McKenna, M.; Lyumkis, D. Sub-2 Å Ewald curvature corrected structure of an AAV2 capsid variant. Nat. Commun. 2018, 9, 3628. [Google Scholar] [CrossRef]
- Meyer, N.L.; Hu, G.; Davulcu, O.; Xie, Q.; Noble, A.J.; Yoshioka, C.; Gingerich, D.S.; Trzynka, A.; David, L.; Stagg, S.M.; et al. Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Zabaleta, N.; Zinn, E.; Pillay, S.; Zengel, J.; Porter, C.; Franceschini, J.S.; Estelien, R.; Carette, J.E.; Zhou, G.L.; et al. GPR108 is a highly conserved AAV entry factor. Mol. Ther. 2020, 28, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.S.; Enderlein, D.; Nelson, C.D.; Carter, W.S.; Kawano, M.; Xing, L.; Swenson, R.D.; Olson, N.H.; Baker, T.S.; Cheng, R.H.; et al. The structure of avian polyomavirus reveals variably sized capsids, non-conserved inter-capsomere interactions, and a possible location of the minor capsid protein VP4. Virology 2011, 411, 142–152. [Google Scholar] [CrossRef]
- Neu, U.; Woellner, K.; Gauglitz, G.; Stehle, T. Structural basis of GM1 ganglioside recognition by simian virus 40. Proc. Natl. Acad. Sci. USA 2008, 105, 5219–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Aronovich, E.L.; McIvor, R.S.; Hackett, P.B. The Sleeping Beauty transposon system: A non-viral vector for gene therapy. Hum. Mol. Genet. 2011, 20, R14–R20. [Google Scholar] [CrossRef] [Green Version]
- Vigdal, T.J.; Kaufman, C.D.; Izsvák, Z.; Voytas, D.F.; Ivics, Z. Common physical properties of DNA affecting target site selection of Sleeping Beauty and other Tc1/mariner transposable elements. J. Mol. Biol. 2002, 323, 441–452. [Google Scholar] [CrossRef]
- Liu, G.; Geurts, A.M.; Yae, K.; Srinivasan, A.R.; Fahrenkrug, S.C.; Largaespada, D.A.; Takeda, J.; Horie, K.; Olson, W.K.; Hackett, P.B. Target-site preferences of Sleeping Beauty transposons. J. Mol. Biol. 2005, 346, 161–173. [Google Scholar] [CrossRef]
- Gogol-Döring, A.; Ammar, I.; Gupta, S.; Bunse, M.; Miskey, C.; Chen, W.; Uckert, W.; Schulz, T.F.; Izsvák, Z.; Ivics, Z. Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the piggyBac transposon in primary human CD4(+) T cells. Mol. Ther. 2016, 24, 592–606. [Google Scholar] [CrossRef] [Green Version]
- Fernando, S.; Fletcher, B.S. Sleeping Beauty transposon-mediated nonviral gene therapy. BioDrugs 2006, 20, 219–229. [Google Scholar] [CrossRef]
- Hackett, P.B.; Largaespada, D.A.; Cooper, L.J. A transposon and transposase system for human application. Mol. Ther. 2010, 18, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Izsvák, Z. Transposons for gene therapy! Curr. Gene Ther. 2006, 6, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Izsvák, Z.; Hackett, P.B.; Cooper, L.J.; Ivics, Z. Translating Sleeping Beauty transposition into cellular therapies: Victories and challenges. Bioessays 2010, 32, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeb, K.R.; Hughes, B.T.; Fissel, B.M.; Osteen, N.J.; Knoblaugh, S.E.; Grim, J.E.; Drury, L.J.; Sarver, A.; Dupuy, A.J.; Clurman, B.E. Insertional mutagenesis using the Sleeping Beauty transposon system identifies drivers of erythroleukemia in mice. Sci. Rep. 2019, 9, 5488. [Google Scholar] [CrossRef]
- Hackett, P.B.; Largaespada, D.A.; Switzer, K.C.; Cooper, L.J. Evaluating risks of insertional mutagenesis by DNA transposons in gene therapy. Transl. Res. 2013, 161, 265–283. [Google Scholar] [CrossRef] [Green Version]
- Aravalli, R.N.; Belcher, J.D.; Steer, C.J. Liver-targeted gene therapy: Approaches and challenges. Liver Transpl. 2015, 21, 718–737. [Google Scholar] [CrossRef] [Green Version]
- Aravalli, R.N.; Steer, C.J. Gene editing technology as an approach to the treatment of liver diseases. Expert Opin. Biol. Ther. 2016, 16, 595–608. [Google Scholar] [CrossRef]
- Zabaleta, N.; Hommel, M.; Salas, D.; Gonzalez-Aseguinolaza, G. Genetic-based approaches to inherited metabolic liver diseases. Hum. Gene Ther. 2019, 30, 1190–1203. [Google Scholar] [CrossRef]
- Turunen, T.A.; Kurkipuro, J.; Heikura, T.; Vuorio, T.; Hytönen, E.; Izsvák, Z.; Ylä-Herttuala, S. Sleeping Beauty transposon vectors in liver-directed gene delivery of LDLR and VLDLR for gene therapy of familial hypercholesterolemia. Mol. Ther. 2016, 24, 620–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portier, I.; Vanhoorelbeke, K.; Verhenne, S.; Pareyn, I.; Vandeputte, N.; Deckmyn, H.; Goldenberg, D.S.; Samal, H.B.; Singh, M.; Ivics, Z.; et al. High and long-term von Willebrand factor expression after Sleeping Beauty transposon-mediated gene therapy in a mouse model of severe von Willebrand disease. J. Thromb. Haemost. 2018, 16, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, M.J.; Smith, G.E.; Summers, M.D. Acquisition of host cell DNA sequences by baculoviruses: Relationship between host DNA insertions and FP mutants of Autographa californica and Galleria mellonella nuclear polyhedrosis viruses. J. Virol. 1983, 47, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cary, L.C.; Goebel, M.; Corsaro, B.G.; Wang, H.G.; Rosen, E.; Fraser, M.J. Transposon mutagenesis of baculoviruses: Analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology 1989, 172, 156–169. [Google Scholar] [CrossRef]
- Li, M.A.; Turner, D.J.; Ning, Z.; Yusa, K.; Liang, Q.; Eckert, S.; Rad, L.; Fitzgerald, T.W.; Craig, N.L.; Bradley, A. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res. 2011, 39, e148. [Google Scholar] [CrossRef] [Green Version]
- Elick, T.A.; Bauser, C.A.; Fraser, M.J. Excision of the piggyBac transposable element in vitro is a precise event that is enhanced by the expression of its encoded transposase. Genetica 1996, 98, 33–41. [Google Scholar] [CrossRef]
- Saridey, S.K.; Liu, L.; Doherty, J.E.; Kaja, A.; Galvan, D.L.; Fletcher, B.S.; Wilson, M.H. PiggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol. Ther. 2009, 17, 2115–2120. [Google Scholar] [CrossRef]
- Nakanishi, H.; Higuchi, Y.; Kawakami, S.; Yamashita, F.; Hashida, M. PiggyBac transposon-mediated long-term gene expression in mice. Mol. Ther. 2010, 18, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Fujimoto, N.; Sasakawa, N.; Ohinata, Y.; Shima, M.; Yamanaka, S.; Sugimoto, M.; Hotta, A. Delivery of full-length factor VIII using a piggyBac transposon vector to correct a mouse model of hemophilia A. PLoS ONE 2014, 9, e104957. [Google Scholar] [CrossRef] [Green Version]
- Di Matteo, M.; Samara-Kuko, E.; Ward, N.J.; Waddington, S.N.; Waddingon, S.N.; McVey, J.H.; Chuah, M.K.; VandenDriessche, T. Hyperactive piggyBac transposons for sustained and robust liver-targeted gene therapy. Mol. Ther. 2014, 22, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Guo, H.; Tammana, S.; Jung, Y.C.; Mellgren, E.; Bassi, P.; Cao, Q.; Tu, Z.J.; Kim, Y.C.; Ekker, S.C.; et al. Gene transfer efficiency and genome-wide integration profiling of Sleeping Beauty, Tol2, and piggyBac transposons in human primary T cells. Mol. Ther. 2010, 18, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Mehier-Humbert, S.; Guy, R.H. Physical methods for gene transfer: Improving the kinetics of gene delivery into cells. Adv. Drug Deliv. Rev. 2005, 57, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.J. Gene therapy progress and prospects: Electroporation and other physical methods. Gene Ther. 2004, 11, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, C.M.; Bettinger, T. Gene therapy progress and prospects: Ultrasound for gene transfer. Gene Ther. 2007, 14, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Plank, C.; Schillinger, U.; Scherer, F.; Bergemann, C.; Rémy, J.S.; Krötz, F.; Anton, M.; Lausier, J.; Rosenecker, J. The magnetofection method: Using magnetic force to enhance gene delivery. Biol. Chem. 2003, 384, 737–747. [Google Scholar] [CrossRef]
- Zhang, G.; Budker, V.G.; Ludtke, J.J.; Wolff, J.A. Naked DNA gene transfer in mammalian cells. Methods Mol. Biol. 2004, 245, 251–264. [Google Scholar] [CrossRef]
- Kawabata, K.; Takakura, Y.; Hashida, M. The fate of plasmid DNA after intravenous injection in mice: Involvement of scavenger receptors in its hepatic uptake. Pharm. Res. 1995, 12, 825–830. [Google Scholar] [CrossRef]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2009, 109, 259–302. [Google Scholar] [CrossRef]
- Li, W.; Szoka, F.C. Lipid-based nanoparticles for nucleic acid delivery. Pharm. Res. 2007, 24, 438–449. [Google Scholar] [CrossRef]
- Sokolova, V.; Epple, M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew. Chem. Int. Ed. Engl. 2008, 47, 1382–1395. [Google Scholar] [CrossRef]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Klibanov, A.M. Non-viral gene therapy: Polycation-mediated DNA delivery. Appl. Microbiol. Biotechnol. 2003, 62, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef]
- Martin, M.E.; Rice, K.G. Peptide-guided gene delivery. AAPS J. 2007, 9, E18–E29. [Google Scholar] [CrossRef] [Green Version]
- Morille, M.; Passirani, C.; Vonarbourg, A.; Clavreul, A.; Benoit, J.P. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials 2008, 29, 3477–3496. [Google Scholar] [CrossRef] [Green Version]
- Cohen, R.N.; van der Aa, M.A.; Macaraeg, N.; Lee, A.P.; Szoka, F.C. Quantification of plasmid DNA copies in the nucleus after lipoplex and polyplex transfection. J. Control. Release 2009, 135, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Sardh, E.; Harper, P.; Balwani, M.; Stein, P.; Rees, D.; Bissell, D.M.; Desnick, R.; Parker, C.; Phillips, J.; Bonkovsky, H.L.; et al. Phase 1 trial of an RNA interference therapy for acute intermittent porphyria. N. Engl. J. Med. 2019, 380, 549–558. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- McGregor, T.L.; Hunt, K.A.; Yee, E.; Mason, D.; Nioi, P.; Ticau, S.; Pelosi, M.; Loken, P.R.; Finer, S.; Lawlor, D.A.; et al. Characterising a healthy adult with a rare HAO1 knockout to support a therapeutic strategy for primary hyperoxaluria. Elife 2020, 9. [Google Scholar] [CrossRef]
- Machin, N.; Ragni, M.V. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J. Blood Med. 2018, 9, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javanbakht, H.; Mueller, H.; Walther, J.; Zhou, X.; Lopez, A.; Pattupara, T.; Blaising, J.; Pedersen, L.; Albæk, N.; Jackerott, M.; et al. Liver-targeted anti-HBV single-stranded oligonucleotides with locked nucleic acid potently reduce HBV gene expression in vivo. Mol. Ther. Nucleic Acids 2018, 11, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, B.; Allegri, G.; Liu, X.B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Häberle, J.; Martini, P.G.V.; Lipshutz, G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar] [CrossRef]
- Jiang, J.; Gusev, Y.; Aderca, I.; Mettler, T.A.; Nagorney, D.M.; Brackett, D.J.; Roberts, L.R.; Schmittgen, T.D. Association of microRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin. Cancer Res. 2008, 14, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Ura, S.; Honda, M.; Yamashita, T.; Ueda, T.; Takatori, H.; Nishino, R.; Sunakozaka, H.; Sakai, Y.; Horimoto, K.; Kaneko, S. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology 2009, 49, 1098–1112. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Kutay, H.; Bai, S.; Datta, J.; Motiwala, T.; Pogribny, I.; Frankel, W.; Jacob, S.T.; Ghoshal, K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J. Cell. Biochem. 2006, 99, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.C.; Hsu, P.W.; Lai, T.C.; Chau, G.Y.; Lin, C.W.; Chen, C.M.; Lin, C.D.; Liao, Y.L.; Wang, J.L.; Chau, Y.P.; et al. microRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology 2009, 49, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schüttler, C.G.; Fehr, C.; Jünemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Chen, J.; Huang, Z. Recent progress in microRNA-based delivery systems for the treatment of human disease. ExRNA 2019, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef]
- Li, H.; Haurigot, V.; Doyon, Y.; Li, T.; Wong, S.Y.; Bhagwat, A.S.; Malani, N.; Anguela, X.M.; Sharma, R.; Ivanciu, L.; et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 2011, 475, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Anguela, X.M.; Sharma, R.; Doyon, Y.; Miller, J.C.; Li, H.; Haurigot, V.; Rohde, M.E.; Wong, S.Y.; Davidson, R.J.; Zhou, S.; et al. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 2013, 122, 3283–3287. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015, 126, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Porro, F.; Bockor, L.; De Caneva, A.; Bortolussi, G.; Muro, A.F. Generation of Ugt1-deficient murine liver cell lines using TALEN technology. PLoS ONE 2014, 9, e104816. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.S.; Murayama, A.; Kang, W.; Kim, M.S.; Yoon, S.K.; Fukasawa, M.; Kondoh, M.; Kim, J.S.; Kim, H.; Kato, T.; et al. Hepatitis C virus entry is impaired by claudin-1 downregulation in diacylglycerol acyltransferase-1-deficient cells. J. Virol. 2014, 88, 9233–9244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Luo, M.; Hayes, R.P.; Kim, J.; Ng, S.; Ding, F.; Liao, M.; Ke, A. Structure basis for directional R-loop formation and substrate handover mechanisms in type I CRISPR-Cas system. Cell 2017, 170, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, K.E.; Lee, C.M.; Yeh, Y.H.; Hsu, R.H.; Gupta, R.; Zhang, M.; Rodriguez, P.J.; Lee, C.S.; Gillard, B.K.; Bissig, K.D.; et al. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 2017, 7, 44624. [Google Scholar] [CrossRef] [Green Version]
- Sin, Y.Y.; Price, P.R.; Ballantyne, L.L.; Funk, C.D. Proof-of-concept gene editing for the murine model of inducible arginase-1 deficiency. Sci. Rep. 2017, 7, 2585. [Google Scholar] [CrossRef] [Green Version]
- Stephens, C.J.; Kashentseva, E.; Everett, W.; Kaliberova, L.; Curiel, D.T. Targeted in vivo knock-in of human alpha-1-antitrypsin cDNA using adenoviral delivery of CRISPR/Cas9. Gene Ther. 2018, 25, 139–156. [Google Scholar] [CrossRef] [Green Version]
- Estève, J.; Blouin, J.M.; Lalanne, M.; Azzi-Martin, L.; Dubus, P.; Bidet, A.; Harambat, J.; Llanas, B.; Moranvillier, I.; Bedel, A.; et al. Generation of induced pluripotent stem cells-derived hepatocyte-like cells for ex vivo gene therapy of primary hyperoxaluria type 1. Stem Cell Res. 2019, 38, 101467. [Google Scholar] [CrossRef] [PubMed]
- Estève, J.; Blouin, J.M.; Lalanne, M.; Azzi-Martin, L.; Dubus, P.; Bidet, A.; Harambat, J.; Llanas, B.; Moranvillier, I.; Bedel, A.; et al. Targeted gene therapy in human-induced pluripotent stem cells from a patient with primary hyperoxaluria type 1 using CRISPR/Cas9 technology. Biochem. Biophys. Res. Commun. 2019, 517, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bai, L.; Zheng, S.; Liu, M.; Zhang, J.; Wang, T.; Xu, Z.; Chen, Y.; Li, J.; Duan, Z. Efficient inhibition of duck hepatitis B virus DNA by the CRISPR/Cas9 system. Mol. Med. Rep. 2017, 16, 7199–7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyushev, D.; Brezgin, S.; Kostyusheva, A.; Zarifyan, D.; Goptar, I.; Chulanov, V. Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell. Mol. Life Sci. 2019, 76, 1779–1794. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, M.; Gong, M.; Xu, Y.; Xie, C.; Deng, H.; Li, X.; Wu, H.; Wang, Z. Inhibition of hepatitis B virus replication via HBV DNA cleavage by Cas9 from Staphylococcus aureus. Antivir. Res. 2018, 152, 58–67. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qi, X.; Zeng, Z.; Wang, L.; Wang, J.; Zhang, T.; Xu, Q.; Shen, C.; Zhou, G.; Yang, S.; et al. CRISPR/Cas9-mediated p53 and Pten dual mutation accelerates hepatocarcinogenesis in adult hepatitis B virus transgenic mice. Sci. Rep. 2017, 7, 2796. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Häberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019. [Google Scholar] [CrossRef]
- Chung, S.J.; Fromme, J.C.; Verdine, G.L. Structure of human cytidine deaminase bound to a potent inhibitor. J. Med. Chem. 2005, 48, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Arvai, A.S.; Sanderson, R.J.; Slupphaug, G.; Kavli, B.; Krokan, H.E.; Mosbaugh, D.W.; Tainer, J.A. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: Protein mimicry of DNA. Cell 1995, 82, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, M.M.; Jessen, S.M.; Ogata, C.M.; Telesnitsky, A.; Goff, S.P.; Hendrickson, W.A. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure 1995, 3, 879–892. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Vector | Payload | Disorder | Preclinical Results/Clinical Trial Endpoints | Clinical Trial/Phase | References |
|---|---|---|---|---|---|
| Retroviral | Factor VIII | Hemophilia A | Physiologic FVIII levels (mice) | [6] | |
| Lentiviral | Factor IX | Hemophilia B | Supraphysiologic FIX levels (nonhuman primates) | [20] | |
| Lentiviral | Factor IX | Hemophilia B | Sustained FIX expression, prevention of NAb induction (mice) | [21] | |
| Lentiviral | CRISPR/Cas9 | Hepatitis B | Inhibition of HBV replication in vivo (mice) and in vitro | [22,23] | |
| Lentiviral | Factor IX | Hemophilia B | Ex vivo stem cell gene correction and autologous stem cell transplant | NCT03961243/Phase I | |
| Adenoviral | UGT1A1 | Crigler-Najjar type I | Correction of hyperbilirubinemia (rats) | [26] | |
| Adenoviral | Human aquaporin 1 | Estrogen-induced cholestasis | Lower serum bile salt concentration, improved biliary output (rats) | [27] | |
| Adenoviral | miR-221/222 | NASH | Decreased hepatic fibrosis (mice) | [28] | |
| HDAd/Sleeping Beauty | Factor IX | Hemophilia B | Stable FIX expression (dogs) | [39,40] | |
| HDAd | AGT | Primary hyperoxaluria 1 | Stable transgene expression, decreased hyperoxaluria (mice) | [41] | |
| HDAd | AAT | AAT deficiency | Stable AAT expression (nonhuman primates) | [32] | |
| AAV | Factor IX | Hemophilia B | FIX expression decreased by 92% following 2/3 hepatectomy (mice) | [48] | |
| AAV | Factor IX | Hemophilia B | Robust FIX expression levels (mice) | [49] | |
| AAV | Copper transporting ATPase 2 | Wilson’s disease | Normalized serum holoceruloplasmin levels and hepatic parenchymal copper levels (mice) | [58] | |
| AAV | Human porphobilinogen | Acute intermittent porphyria | Reduced frequency of biochemical attacks, degree of neuropathy (mice) | [59,60] | |
| AAV | Propionyl-CoA | Propionyl acidemia | Decreased serum toxin level (mice) | [61] | |
| AAV | Human methylmalonyl-CoA mutase | Methylmalonic acidemia | Correction of acidemia, prevention of murine neonatal lethality | [62] | |
| AAV | Phenylalanine hydroxylase | Phenylketonuria | Reduced serum phenylalanine levels (mice) | [63] | |
| AAV | Glucose-6-phosphatase | Glycogen storage disease type Ia | Stable G6P expression, correction of hypoglycemia, normalized hepatic glycogen and reduced hepatic steatosis (mice, dogs) | [64,65] | |
| AAV | ABCB4 | Progressive familial intrahepatic cholestasis 3 | Stable ABCB4 expression, reduced progression of liver fibrosis (mice) | [66] | |
| AAV | Factor VIII | Hemophilia A | Stable FVIII expression (dogs) | [67,68] | |
| AAV | Factor IX | Hemophilia B | Stable FIX expression | NCT00979238/Phase I | [69] |
| AAV | Factor VIII | Hemophilia A | Stable FVIII expression | NCT02576795/Phase I/II | [45] |
| AAV | Factor IX | Hemophilia B | Stable FIX expression | NCT02484092/Phase II | [70,71] |
| AAV | Porphobilinogen deaminase | Acute intermittent porphyria | Safety, not metabolic correction at doses tested, varied results | NCT02082860/Phase I | [72] |
| AAV | LDLR | Homozygous familial hypercholesterolemia | Improvement of lipid profile | NCT02651675/Phase I/II | |
| AAV | Padua variant factor IX | Hemophilia B | Supraphysiologic FIX expression level | NCT03569891/Phase III | |
| AAV | Fidanacogene elaparvovec (high activity factor IX) | Hemophilia B | Supraphysiologic FIX expression level | NCT03587116/Phase III | |
| AAV | GS010 (human wild-type ND4) | Leber hereditary optic neuropathy | Recovery of vision | NCT02652780 NCT02652767 NCT03293524, all Phase III | |
| AAV | LYS-SAF302 (N-sulfoglucosamine sulfohydrolase) | Mucopolysaccharidosis IIIA | Improvement or stabilization of neurodevelopmental state | NCT03612869/Phase III | |
| AAV | NSR-REP1 (REP1) | Choroideremia | Improvement in best corrected visual acuity | NCT03496012/Phase III | |
| AAV | Valoctocogene roxaparvovec (factor VIII) | Hemophilia A | Improvement in FVIII median activity | NCT03370913 NCT03392974, both Phase III | |
| AAV | Voretigene neparvovec-rzyl (RPE65) | Biallelic RPE65 mutation-associated retinal dystrophy | Improvement in multi-lumen mobility test scores | FDA approved | [73] |
| AAV | Onasemnogene abeparvovec-xioi (SMN) | Spinal muscular atrophy | Prevention of death and permanent breathing support | FDA approved | [74] |
| SV40 | UGT1A1 | Crigler-Najjar type I | Normalization of murine ALT and bilirubin serum levels, liver histology | [82] | |
| Sleeping Beauty | Factor VIII | Hemophilia A | Stable FVIII expression | [108,109,110] | |
| Sleeping Beauty | Factor IX | Hemophilia B | Stable FIX expression | [108,109,110] | |
| Sleeping Beauty | AAT | AAT deficiency | Stable AAT expression (mice) | [108,109,110] | |
| Sleeping Beauty | β-glucuronidase | Mucopolysaccharidosis type VII | Stable β-glucuronidase expression (mice) | [108,109,110] | |
| Sleeping Beauty | α-L-iduronidase | Mucopolysaccharidosis type I | Stable α-L-iduronidase expression (mice) | [108,109,110] | |
| Sleeping Beauty | Fah | Hereditary tyrosinemia | Stable Fah expression (mice) | [108,109,110] | |
| Sleeping Beauty | LDLR, VLDLR | Familial hypercholesterolemia | Moderate reduction in plasma cholesterol and atherosclerosis (mice) | [111] | |
| Sleeping Beauty | von Willebrand factor | von Willebrand disease | Supraphysiologic VFW levels (mice) | [112] | |
| piggyBac | Factor VIII | Hemophilia A | Stable FVIII expression | [119] | |
| piggyBac | Factor IX | Hemophilia B | Stable FIX expression | [120] | |
| Non-viral (trivalent N-acetylgalactosamine) | Givosiran (siRNA) | Acute intermittent porphyria | Silences δ-aminolevulinic acid synthase 1 mRNA, reduces AIP attack frequency | FDA approved | [138] |
| Non-viral (liposomes) | Patisiran (siRNA) | Transthyretin-related hereditary amyloidosis | Improvement in polyneuropathy | FDA approved | [139] |
| Non-viral (trivalent N-acetylgalactosamine) | Lumasiran (siRNA) | Primary hyperoxaluria 1 | Decreased hyperoxaluria by silencing glycolate oxidase | NCT02706886/Phase I/II | [140] |
| Non-viral (trivalent N-acetylgalactosamine) | Fitusiran (siRNA) | Hemophilia A & B | Reduces bleeding instances by silencing antithrombin | NCT03417245/Phase III | [141] |
| Non-viral (trivalent N-acetylgalactosamine) | ALN-HBV02 (siRNA) | Hepatitis B | Reduction in HBV surface antigen levels | NCT02826018/Phase I | [142] |
| Non-viral (lipid nanoparticle) | Arginase 1 | Arginine deficiency | Stable, moderate arginase 1 expression (mice) | [143] | |
| Non-viral (SQ injection) | miR-122 antagomir | NASH | Improvement in hepatic steatosis and reduction in plasma cholesterol (mice) | [150] | |
| Non-viral (SQ injection) | miR-122 antagomir | Hepatitis C | Inhibition of viral replication and translation in vitro | [149,153] | |
| AAV | miR-26A | Hepatocellular carcinoma | Suppression of tumorigenesis and induction of tumor-specific apoptosis (mice) | [154] | |
| AAV | Factor VIII, ZFN | Hemophilia A | Stable expression of FVIII (mice) | [157] | |
| AAV | Factor IX, ZFN | Hemophilia B | Stable expression of FIX (mice) | [158,159] | |
| AAV | Human α-galactosidase A, ZFN | Fabry disease | Stable expression of α-galactosidase A (mice) | [159] | |
| AAV | Acid β-glucosidase, ZFN | Gaucher disease | Stable expression of acid β-glucosidase (mice) | [159] | |
| AAV | Iduronate-2 sulfatase, ZFN | Hunter syndrome | Stable expression of iduronate-2 sulfatase (mice) | [159] | |
| AAV | α-L-iduronidase, ZFN | Hurler syndrome | Stable expression of α-L-iduronidase (mice) | [159] | |
| Lentiviral | TALENs targeting diacylglycerol acyltransferase-1 | Hepatitis C | Viral entry impaired in a claudin-1 dependent manner in vitro | [161] | |
| Non-viral (ssDNA oligonucleotides) | Fah, CRISPR/Cas9 | Hereditary tyrosinemia | Correction of Fah mutation (mice) | [166] | |
| AAV | CRISPR/Cas9 | Induction of severe hypercholesterolemia and atherosclerosis | Via mutation of LDLR (mice) | [167] | |
| Adenoviral | AAT, CRISPR/Cas9 | AAT deficiency | Somatic murine hepatic incorporation and stable expression of AAT | [168] | |
| AAV | CRISPR/Cas9 | Hepatitis B | Inhibition of genome replication in mice | [173] | |
| Non-viral (DNA plasmids) | CRISPR/Cas9 | Induction of hepatocellular carcinoma | Via mutation of p53 and Pten (mice) | [176] | |
| None | Ex vivo PD-1 knockout in T lymphocytes engineered via CRISPR | Hepatocellular carcinoma | Tumor response rate, progression-free survival | NCT04417764 | |
| AAV | CRISPR/Cas9 | Phenylketonuria | Correction of PKU mutation via base editing (mice) | [179] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moscoso, C.G.; Steer, C.J. The Evolution of Gene Therapy in the Treatment of Metabolic Liver Diseases. Genes 2020, 11, 915. https://doi.org/10.3390/genes11080915
Moscoso CG, Steer CJ. The Evolution of Gene Therapy in the Treatment of Metabolic Liver Diseases. Genes. 2020; 11(8):915. https://doi.org/10.3390/genes11080915
Chicago/Turabian StyleMoscoso, Carlos G., and Clifford J. Steer. 2020. "The Evolution of Gene Therapy in the Treatment of Metabolic Liver Diseases" Genes 11, no. 8: 915. https://doi.org/10.3390/genes11080915
APA StyleMoscoso, C. G., & Steer, C. J. (2020). The Evolution of Gene Therapy in the Treatment of Metabolic Liver Diseases. Genes, 11(8), 915. https://doi.org/10.3390/genes11080915