Mode and Tempo of Microsatellite Evolution across 300 Million Years of Insect Evolution


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
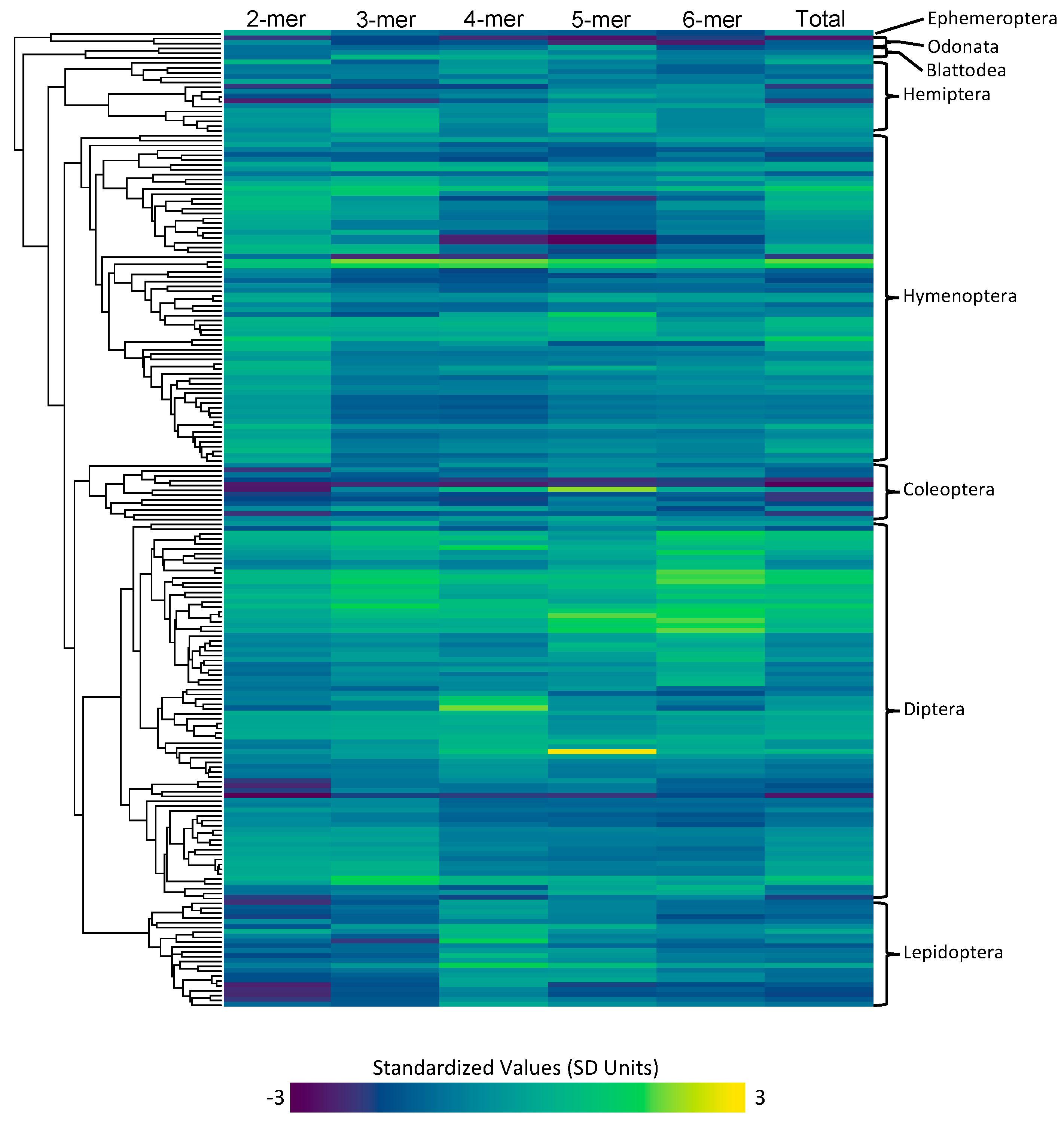
3.1. Data Quality and Collection
3.1.1. Assembly Quality
3.1.2. Phylogenetic Reconstruction
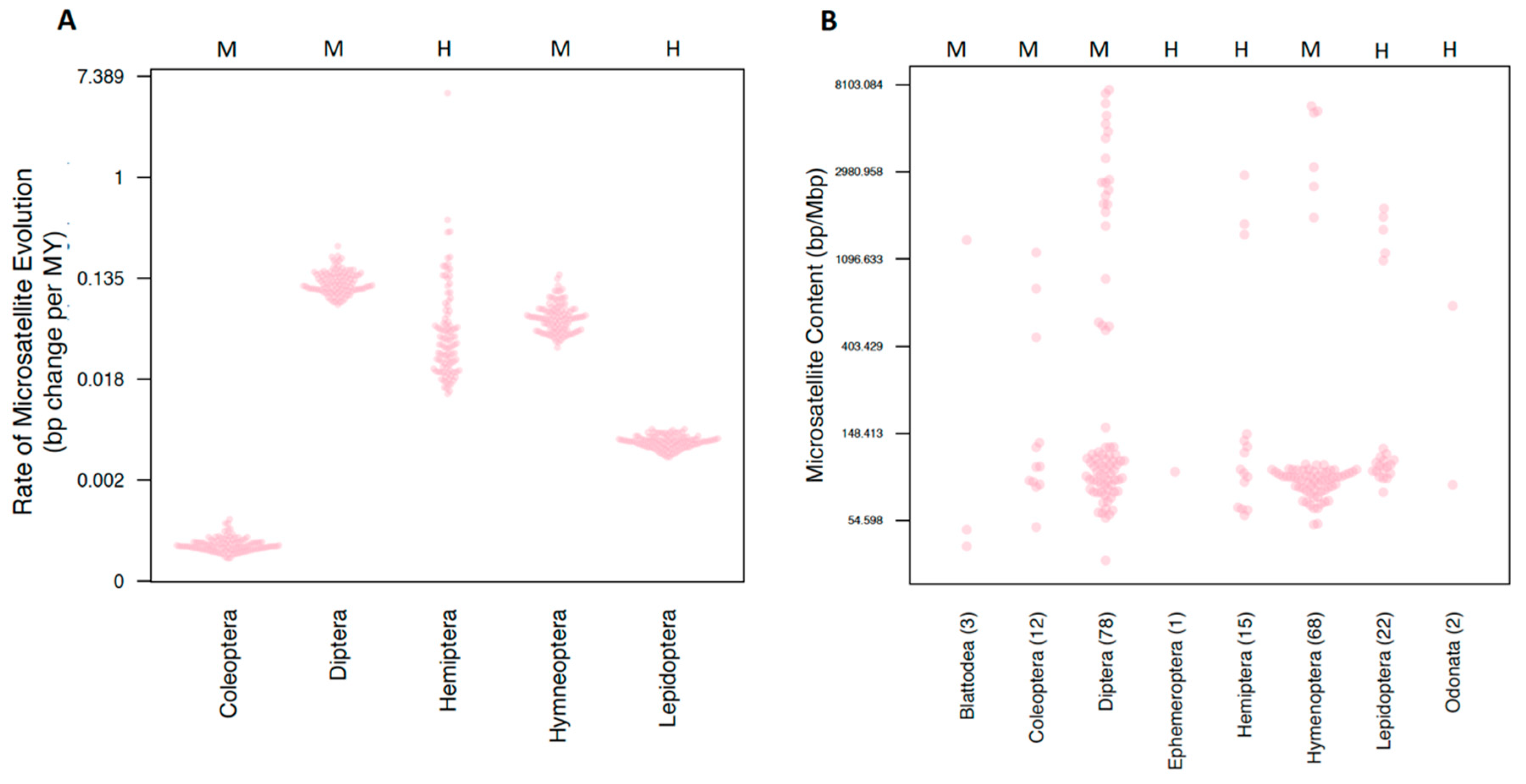
3.1.3. Microsatellite Content and Rates
3.2. Comparative Analyses
3.2.1. Order
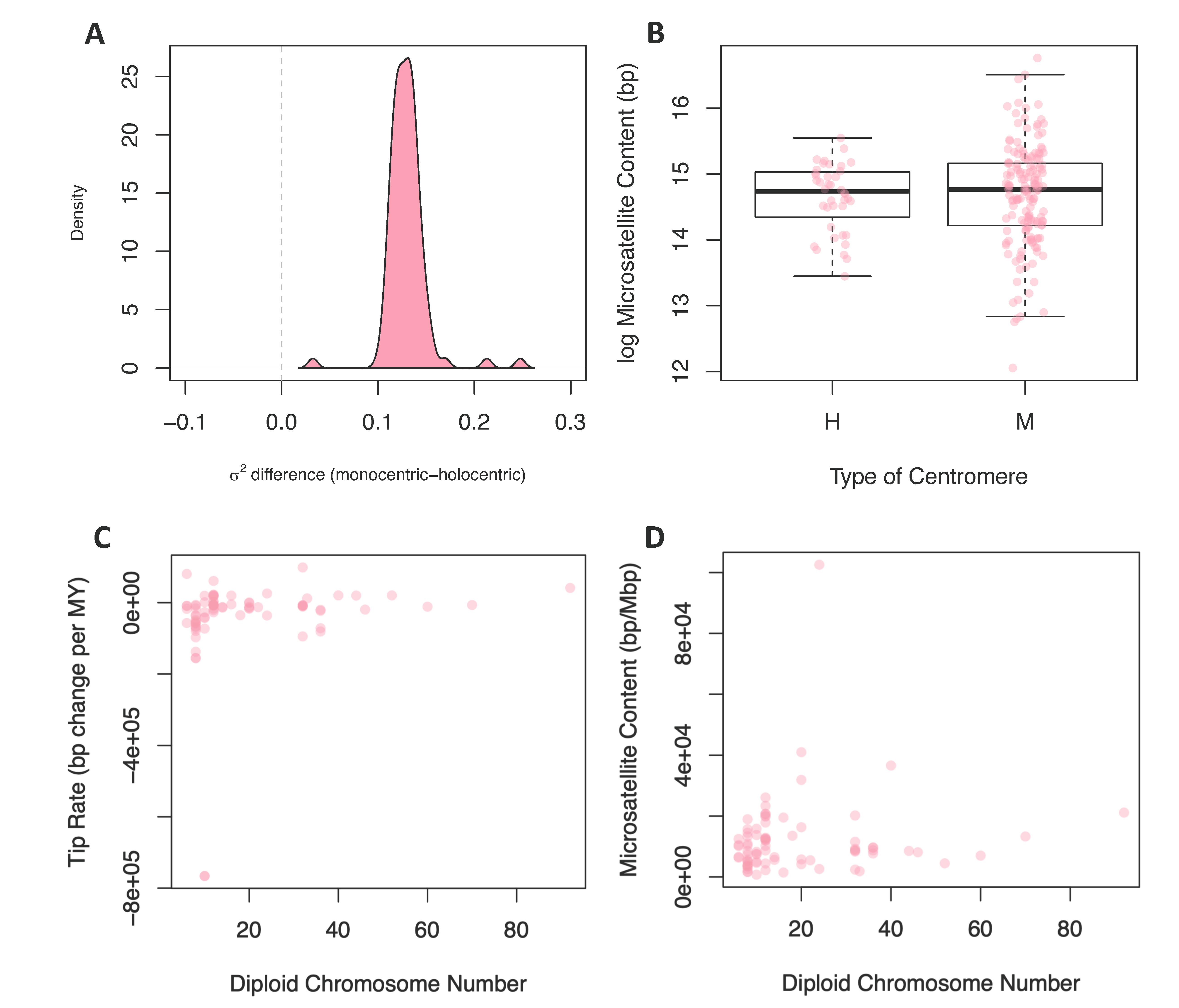
3.2.2. Centromere Type
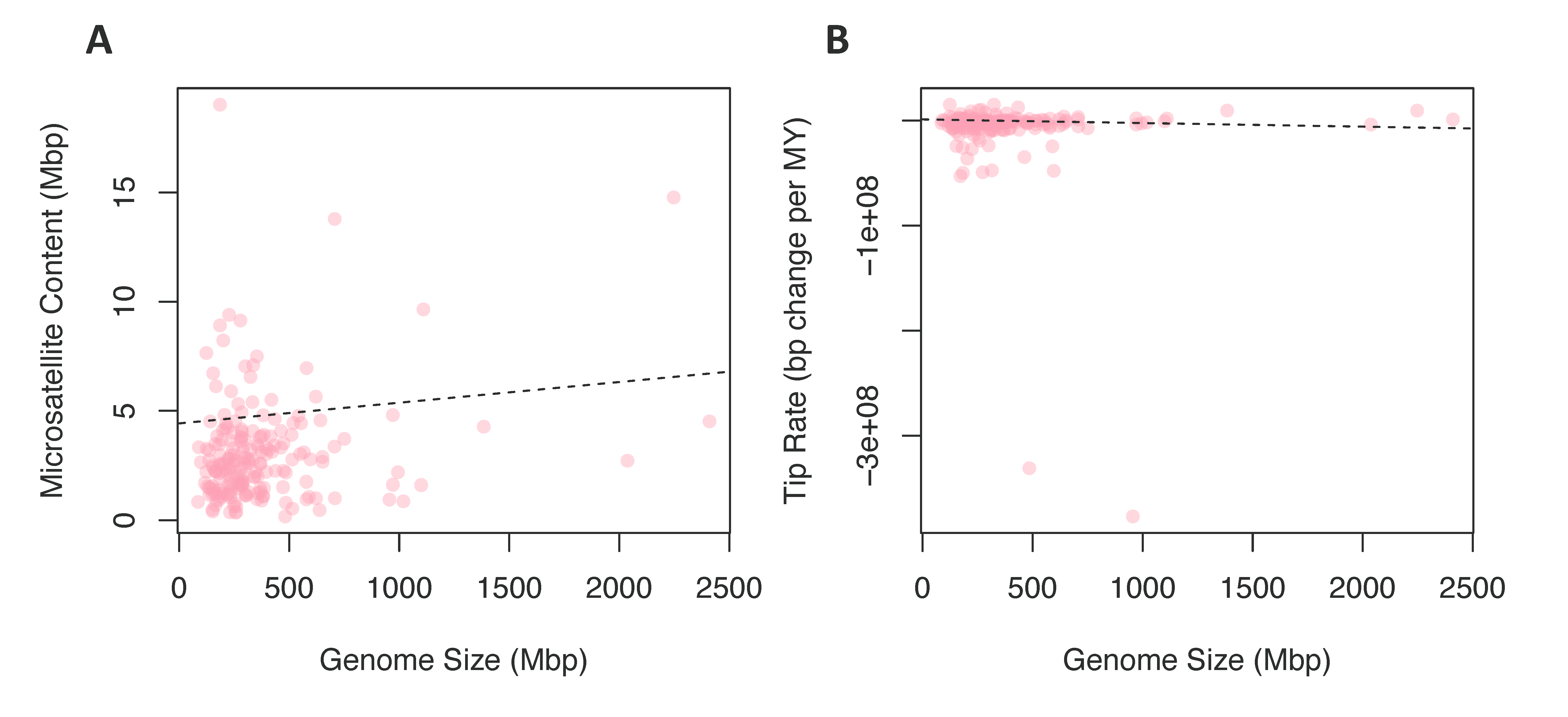
3.2.3. Chromosome Number and Genome Size
4. Discussion
4.1. Traits
4.2. Clades
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar] [PubMed]
- Edwards, Y.J.K.; Elgar, G.; Clark, M.S.; Bishop, M.J. The identification and characterization of microsatellites in the compact genome of the japanese pufferfish, Fugu rubripes: Perspectives in functional and comparative genomic analyses. J. Mol. Biol. 1998, 278, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.; Greenwell, P.W.; Liu, C.-P.; Arnheim, N.; Petes, T.D. Triplet repeats form secondary structures that escape DNA repair in yeast. Proc. Natl. Acad. Sci. USA 1999, 96, 1504. [Google Scholar] [CrossRef] [PubMed]
- Dokholyan, N.V.; Buldyrev, S.V.; Havlin, S.; Stanley, H.E. Distributions of dimeric tandem repeats in non-coding and coding DNA sequences. J. Theor. Biol. 2000, 202, 273–282. [Google Scholar] [CrossRef]
- Zhang, L.; Zuo, K.; Zhang, F.; Cao, Y.; Wang, J.; Zhang, Y.; Sun, X.; Tang, K. Conservation of noncoding microsatellites in plants: Implication for gene regulation. BMC Genom. 2006, 7, 323. [Google Scholar] [CrossRef]
- Fujimori, S.; Washio, T.; Higo, K.; Ohtomo, Y.; Murakami, K.; Matsubara, K.; Kawai, J.; Carninci, P.; Hayashizaki, Y.; Kikuchi, S.; et al. A novel feature of microsatellites in plants: A distribution gradient along the direction of transcription. FEBS Lett. 2003, 554, 17–22. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellite mutations in the germline: Implications for evolutionary inference. Trends Genet. 2000, 16, 551–558. [Google Scholar] [CrossRef]
- Slatkin, M. A measure of population subdivision based on microsatellite allele frequencies. Genetics 1995, 139, 457–462. [Google Scholar]
- Criscione, C.D.; Vilas, R.; Paniagua, E.; Blouin, M.S. More than meets the eye: Detecting cryptic microgeographic population structure in a parasite with a complex life cycle. Mol. Ecol. 2011, 20, 2510–2524. [Google Scholar] [CrossRef]
- Ballantyne, K.N.; Goedbloed, M.; Fang, R.; Schaap, O.; Lao, O.; Wollstein, A.; Choi, Y.; van Duijn, K.; Vermeulen, M.; Brauer, S. Mutability of Y-chromosomal microsatellites: Rates, characteristics, molecular bases, and forensic implications. Am. J. Hum. Genet. 2010, 87, 341–353. [Google Scholar] [CrossRef]
- Blouin, M.S. DNA-based methods for pedigree reconstruction and kinship analysis in natural populations. Trends Ecol. Evol. 2003, 18, 503–511. [Google Scholar] [CrossRef]
- Highnam, G.; Franck, C.; Martin, A.; Stephens, C.; Puthige, A.; Mittelman, D. Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res. 2012, 41, e32. [Google Scholar] [CrossRef]
- Field, D.; Wills, C. Abundant microsatellite polymorphism in Saccharomyces cerevisiae, and the different distributions of microsatellites in eight prokaryotes and S. cerevisiae, result from strong mutation pressures and a variety of selective forces. Proc. Natl. Acad. Sci. USA 1998, 95, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Sinden, R.R. Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry 1996, 35, 5041–5053. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Heslop-Harrison, J.S. The physical and genomic organization of microsatellites in sugar beet. Proc. Natl. Acad. Sci. USA 1996, 93, 8761. [Google Scholar] [CrossRef]
- Hoffman, E.K.; Trusko, S.P.; Murphy, M.; George, D.L. An S1 nuclease-sensitive homopurine/homopyrimidine domain in the c-Ki-ras promoter interacts with a nuclear factor. Proc. Natl. Acad. Sci. USA 1990, 87, 2705. [Google Scholar] [CrossRef]
- Chamberlain, N.L.; Driver, E.D.; Miesfeld, R.L. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res. 1994, 22, 3181–3186. [Google Scholar] [CrossRef]
- Lue, N.F.; Buchman, A.R.; Kornberg, R.D. Activation of yeast RNA polymerase II transcription by a thymidine-rich upstream element in vitro. Proc. Natl. Acad. Sci. USA 1989, 86, 486. [Google Scholar] [CrossRef]
- Sandberg, G.; Schalling, M. Effect of in vitro promoter methylation and CGG repeat expansion on FMR-1 expression. Nucleic Acids Res. 1997, 25, 2883–2887. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Coetzee, G.A.; Irvine, R.A.; Buckley, M.; Ferguson-Smith, M.A. Sequence variation and size ranges of CAG repeats in the Machado-Joseph disease, spinocerebellar ataxia type 1 and androgen receptor genes. Hum. Mol. Genet. 1995, 4, 1585–1590. [Google Scholar] [CrossRef]
- Eisen, J. Mechanistic basis for microsatellite instability. In Microsatellites: Evolution and Applications; Goldstein, D., Schlotterer, C., Eds.; Oxford University Press: Oxford, UK, 1999; pp. 34–48. [Google Scholar]
- Klintschar, M.; Dauber, E.M.; Ricci, U.; Cerri, N.; Immel, U.D.; Kleiber, M.; Mayr, W.R. Haplotype studies support slippage as the mechanism of germline mutations in short tandem repeats. Electrophoresis 2004, 25, 3344–3348. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.J.; Ecker, J.R. Assignment of 30 microsatellite loci to the linkage map of Arabidopsis. Genomics 1994, 19, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Neff, B.D.; Gross, M.R. Microsatellite evolution in vertebrates: Inference from AC dinucleotide repeats. Evolution 2001, 55, 1717–1733. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Blackmon, H.; Reyes-Velasco, J.; Schield, D.R.; Card, D.C.; Andrew, A.; Castoe, T.A. Microsatellite landscape evolutionary dynamics across 450 million years of vertebrate genome evolution. Genome 2016, 59, 295–310. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Thomson, R.C.; Shaffer, H.B. Sparse supermatrices for phylogenetic inference: Taxonomy, alignment, rogue taxa, and the phylogeny of living turtles. Syst. Biol. 2010, 59, 42–58. [Google Scholar] [CrossRef]
- Rabosky, D.L. No substitute for real data: A cautionary note on the use of phylogenies from birth–death polytomy resolvers for downstream comparative analyses. Evolution 2015, 69, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.; Maddison, D. Mesquite: A Modular System for Evolutionary Analysis. 2011. Available online: http://mesquiteproject.org (accessed on 1 November 2019).
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Lo, J.; Jonika, M.M.; Blackmon, H. Microcounter: Microsatellite characterization in genome assemblies. G3 Genes Genomes Genet. 2019, 9, 3101–3104. [Google Scholar] [CrossRef]
- Blackmon, H.; Ross, L.; Bachtrog, D. Sex determination, sex chromosomes, and karyotype evolution in insects. J. Hered. 2017, 108, 78–93. [Google Scholar] [CrossRef]
- The Tree of Sex Consortium. Tree of Sex: A database of sexual systems. Sci. Data 2014, 1, 140015. [Google Scholar] [CrossRef]
- Gregory, T.R. Animal Genome Size Database. 2020. Available online: http://www.genomesize.com (accessed on 1 September 2018).
- Harmon, L.J.; Weir, J.T.; Brock, C.D.; Glor, R.E.; Challenger, W. GEIGER: Investigating evolutionary radiations. Bioinformatics 2008, 24, 129–131. [Google Scholar] [CrossRef]
- Revell, L.J. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Ho, L.S.T.; Ane, C.; Lachlan, R.; Tarpinian, K.; Feldman, R.; Yu, Q.; van der Bijl, W.; Vos R, H.M.L. Package ‘phylolm’. Available online: http://www.cran.r-project.org/web/packages/phylolm/index.html (accessed on 1 February 2018).
- Andy, B.; Mikko, K. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Stephan, W. Recombination and the evolution of satellite DNA. Genet. Res. 1986, 47, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Melters, D.P.; Paliulis, L.V.; Korf, I.F.; Chan, S.W. Holocentric chromosomes: Convergent evolution, meiotic adaptations, and genomic analysis. Chromosome Res. 2012, 20, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Nokkala, S.; Kuznetsova, V.; Maryanska-Nadachowska, A.; Nokkala, C. Holocentric chromosomes in meiosis. I. Restriction of the number of chiasmata in bivalents. Chromosome Res. 2004, 12, 733–739. [Google Scholar] [CrossRef]
- Cuacos, M.; Franklin, H.; Chris, F.; Heckmann, S. Atypical centromeres in plants—What they can tell us. Front. Plant Sci. 2015, 6, 913. [Google Scholar] [CrossRef]
- Mandrioli, M.; Carlo Manicardi, G. Unlocking holocentric chromosomes: New perspectives from comparative and functional genomics? Curr. Genom. 2012, 13, 343–349. [Google Scholar] [CrossRef]
- Heckmann, S.; Macas, J.; Kumke, K.; Fuchs, J.; Schubert, V.; Ma, L.; Novak, P.; Neumann, P.; Taudien, S.; Platzer, M. The holocentric species L uzula elegans shows interplay between centromere and large-scale genome organization. Plant J. 2013, 73, 555–565. [Google Scholar] [CrossRef]
- Subirana, J.A.; Messeguer, X. A satellite explosion in the genome of holocentric nematodes. PLoS ONE 2013, 8, e62221. [Google Scholar] [CrossRef]
- Melters, D.P.; Bradnam, K.R.; Young, H.A.; Telis, N.; May, M.R.; Ruby, J.G.; Sebra, R.; Peluso, P.; Eid, J.; Rank, D.; et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013, 14. [Google Scholar] [CrossRef]
- Rabosky, D.L.; Goldberg, E.E. Model inadequacy and mistaken inferences of trait-dependent speciation. Syst. Biol. 2015, 64, 340–355. [Google Scholar] [CrossRef]
- Bachtrog, D.; Weiss, S.; Zangerl, B.; Brem, G.; Schlötterer, C. Distribution of dinucleotide microsatellites in the Drosophila melanogaster genome. Mol. Biol. Evol. 1999, 16, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Lowenhaupt, K.; Rich, A.; Pardue, M. Nonrandom distribution of long mono-and dinucleotide repeats in Drosophila chromosomes: Correlations with dosage compensation, heterochromatin, and recombination. Mol. Cell. Biol. 1989, 9, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.J.; Yasmineh, W.G. Heterochromatin, satellite DNA, and cell function. Science 1971, 174, 1200–1209. [Google Scholar] [CrossRef]
- Hartl, D.L. Molecular melodies in high and low C. Nat. Rev. Genet. 2000, 1, 145–149. [Google Scholar] [CrossRef]
- Mirsky, A.; Ris, H. The desoxyribonucleic acid content of animal cells and its evolutionary significance. J. Gen. Physiol. 1951, 34, 451. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The origins of genome complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef]
- Petrov, D.A. Evolution of genome size: New approaches to an old problem. TRENDS Genet. 2001, 17, 23–28. [Google Scholar] [CrossRef]
- Kubis, S.; Schmidt, T.; Heslop-Harrison, J.S. Repetitive DNA elements as a major component of plant genomes. Ann. Bot. 1998, 82, 45–55. [Google Scholar] [CrossRef]
- Primmer, C.R.; Raudsepp, T.; Chowdhary, B.P.; Møller, A.P.; Ellegren, H. Low frequency of microsatellites in the avian genome. Genome Res. 1997, 7, 471–482. [Google Scholar] [CrossRef]
- Oliver, M.J.; Petrov, D.; Ackerly, D.; Falkowski, P.; Schofield, O.M. The mode and tempo of genome size evolution in eukaryotes. Genome Res. 2007, 17, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Amos, W.; Leggo, J.; Goodburn, S.; Jain, S.; Li, S.-H.; Margolis, R.L.; Ross, C.A.; Ferguson-Smith, M.A. Microsatellite evolution—Evidence for directionality and variation in rate between species. Nat. Genet. 1995, 10, 337–343. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonika, M.; Lo, J.; Blackmon, H. Mode and Tempo of Microsatellite Evolution across 300 Million Years of Insect Evolution. Genes 2020, 11, 945. https://doi.org/10.3390/genes11080945
Jonika M, Lo J, Blackmon H. Mode and Tempo of Microsatellite Evolution across 300 Million Years of Insect Evolution. Genes. 2020; 11(8):945. https://doi.org/10.3390/genes11080945
Chicago/Turabian StyleJonika, Michelle, Johnathan Lo, and Heath Blackmon. 2020. "Mode and Tempo of Microsatellite Evolution across 300 Million Years of Insect Evolution" Genes 11, no. 8: 945. https://doi.org/10.3390/genes11080945
APA StyleJonika, M., Lo, J., & Blackmon, H. (2020). Mode and Tempo of Microsatellite Evolution across 300 Million Years of Insect Evolution. Genes, 11(8), 945. https://doi.org/10.3390/genes11080945