Manganese Ions Individually Alter the Reverse Transcription Signature of Modified Ribonucleosides

 ,
,  ,
,  and
and
Abstract
:1. Introduction
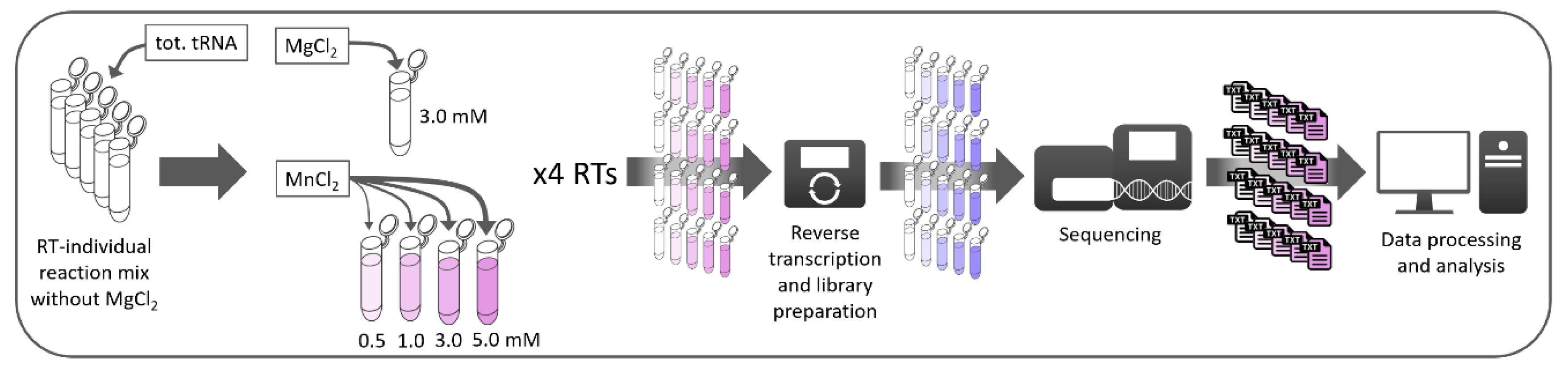
2. Materials and Methods
2.1. Sample Treatment and Library Preparation
2.2. Processing of Sequencing Output
3. Results
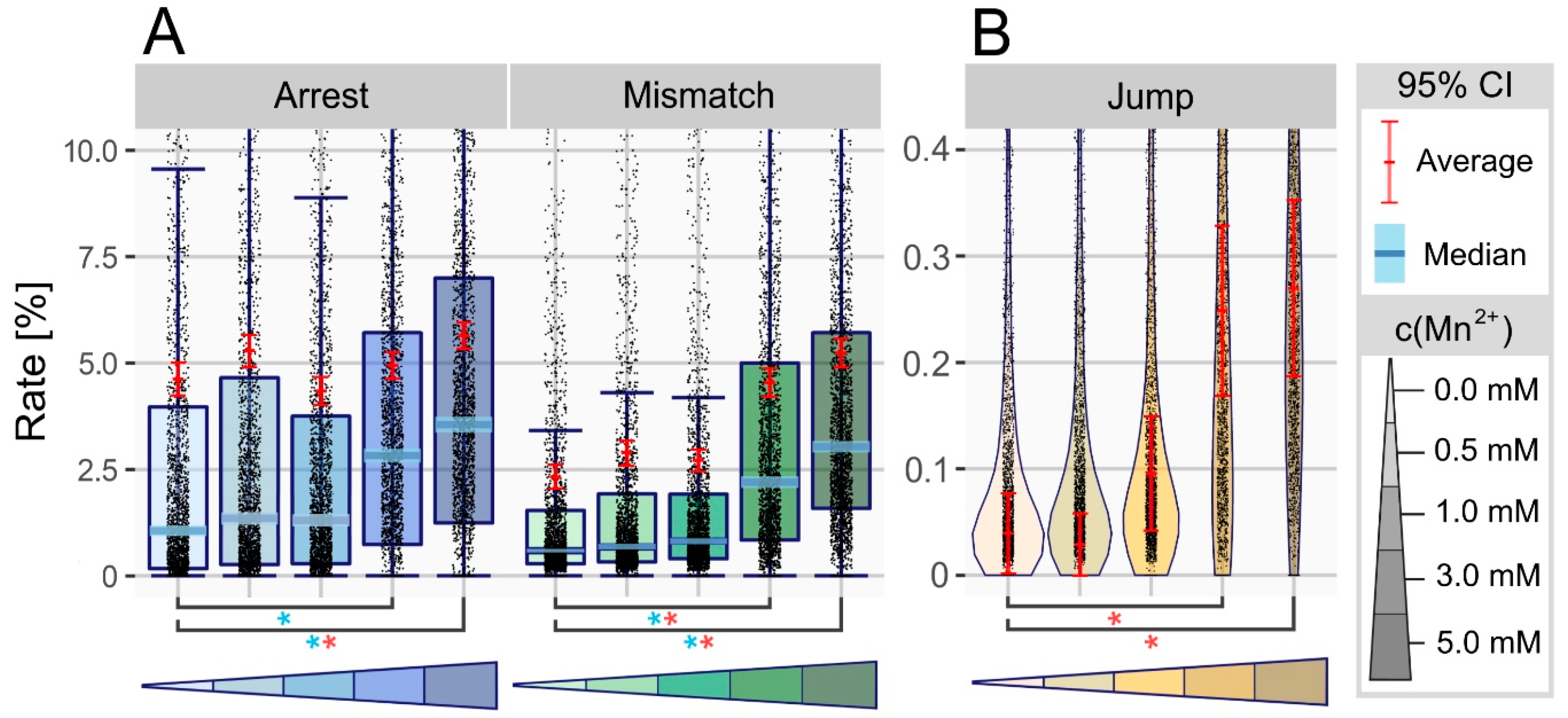
3.1. Manganese Ions Increase Error Rate at Non-Modified Sites
3.2. Manganese Facilitates Overcoming of Blockades in Watson-Crick Base Pairing
3.3. Manganese Enhances the Nucleotide Skipping Ability of Reverse Transcriptases
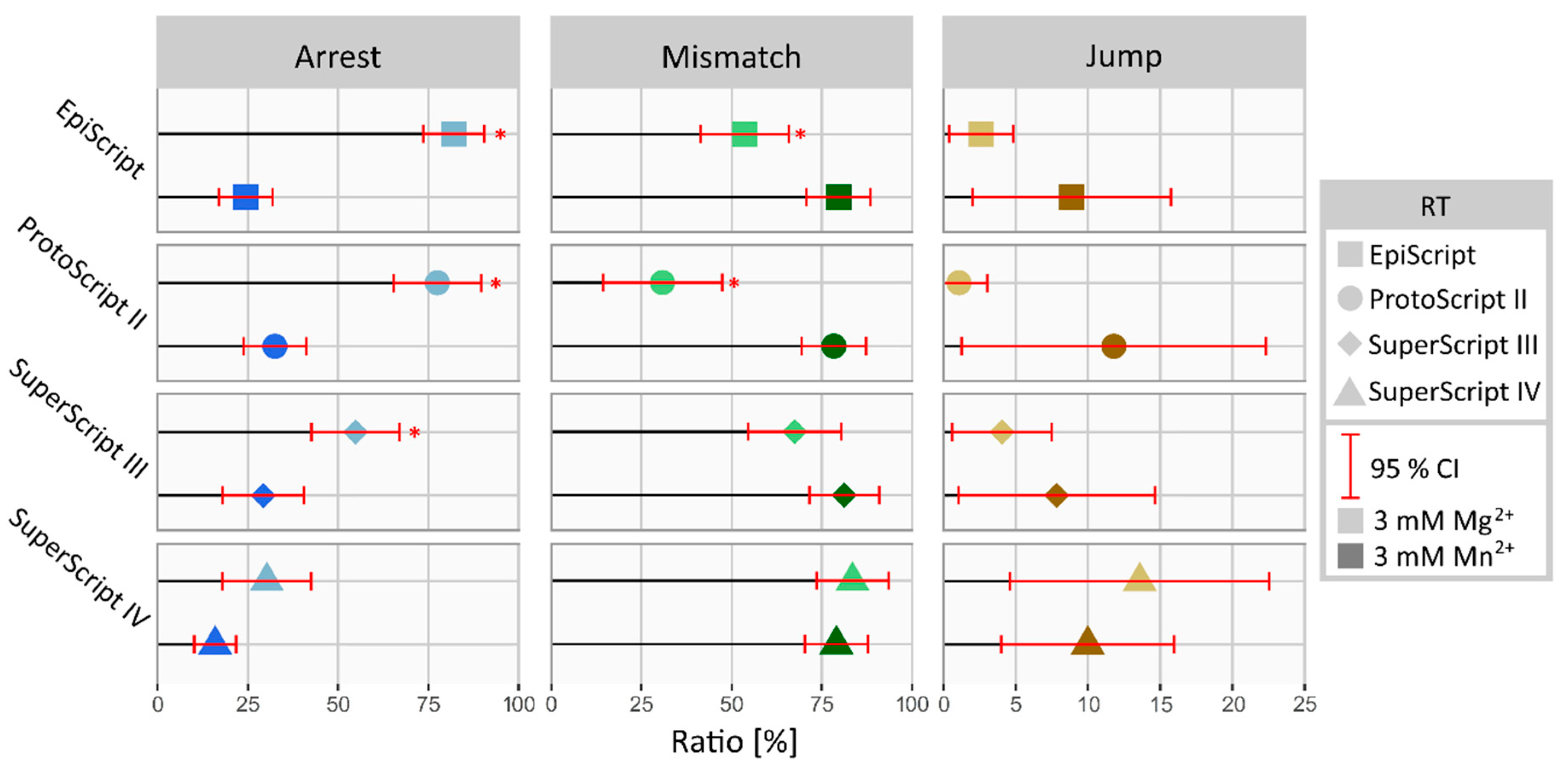
3.4. Manganese-Derived Alteration of RT Performance Is Polymerase-Specific
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Cohn, W.E.; Volkin, E. Nucleoside-5′-Phosphates from Ribonucleic Acid. Nature 1951, 167, 483–484. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Helm, M.; Brule, H.; Degoul, F.; Cepanec, C.; Leroux, J.-P.; Giege, R.; Florentz, C. The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res. 1998, 26, 1636–1643. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Dal Magro, C.; Keller, P.; Kotter, A.; Werner, S.; Duarte, V.; Marchand, V.; Ignarski, M.; Freiwald, A.; Müller, R.-U.; Dieterich, C.; et al. A Vastly Increased Chemical Variety of RNA Modifications Containing a Thioacetal Structure. Angew. Chem. Int. Ed. 2018, 57, 7893–7897. [Google Scholar] [CrossRef]
- Haruehanroengra, P.; Zheng, Y.Y.; Zhou, Y.; Huang, Y.; Sheng, J. RNA modifications and cancer. RNA Biol. 2020. [Google Scholar] [CrossRef]
- Ryvkin, P.; Leung, Y.Y.; Silverman, I.M.; Childress, M.; Valladares, O.; Dragomir, I.; Gregory, B.D.; Wang, L.-S. HAMR: High-throughput annotation of modified ribonucleotides. RNA 2013, 19, 1684–1692. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson, M.; Vollmers, C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauenschild, R.; Werner, S.; Tserovski, L.; Hildebrandt, A.; Motorin, Y.; Helm, M. CoverageAnalyzer (CAn): A Tool for Inspection of Modification Signatures in RNA Sequencing Profiles. Biomolecules 2016, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tserovski, L.; Marchand, V.; Hauenschild, R.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-throughput sequencing for 1-methyladenosine (m1A) mapping in RNA. Methods 2016, 107, 110–121. [Google Scholar] [CrossRef]
- Schmidt, L.; Werner, S.; Kemmer, T.; Niebler, S.; Kristen, M.; Ayadi, L.; Johe, P.; Marchand, V.; Schirmeister, T.; Motorin, Y.; et al. Graphical Workflow System for Modification Calling by Machine Learning of Reverse Transcription Signatures. Front. Genet. 2019, 10, 876. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Schmidt, L.; Marchand, V.; Kemmer, T.; Falschlunger, C.; Sednev, M.V.; Bec, G.; Ennifar, E.; Höbartner, C.; Micura, R.; et al. Machine learning of reverse transcription signatures of variegated polymerases allows mapping and discrimination of methylated purines in limited transcriptomes. Nucleic Acids Res. 2020, 48, 3734–3746. [Google Scholar] [CrossRef] [Green Version]
- Dai, Q.; Zheng, G.; Schwartz, M.H.; Clark, W.C.; Pan, T. Selective Enzymatic Demethylation of N 2, N 2-Dimethylguanosine in RNA and Its Application in High-Throughput tRNA Sequencing. Angew. Chem. Int. Ed. 2017, 56, 5017–5020. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-wide mapping of pseudouridines: Pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE 2014, 9, e110799. [Google Scholar] [CrossRef] [Green Version]
- Marchand, V.; Ayadi, L.; Ernst, F.G.M.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.J.; Motorin, Y. AlkAniline-Seq: Profiling of m 7 G and m 3 C RNA Modifications at Single Nucleotide Resolution. Angew. Chem. Int. Ed. 2018, 57, 16785–16790. [Google Scholar] [CrossRef]
- Schaefer, M. RNA 5-Methylcytosine Analysis by Bisulfite Sequencing. Methods Enzymol. 2015, 560, 297–329. [Google Scholar] [PubMed]
- Hauenschild, R.; Tserovski, L.; Schmid, K.; Thüring, K.; Winz, M.-L.; Sharma, S.; Entian, K.-D.; Wacheul, L.; Lafontaine, D.L.J.; Anderson, J.; et al. The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res. 2015, 43, 9950–9964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motorin, Y.; Helm, M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes (Basel) 2019, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, D.; Krawczak, M. Mechanisms of insertional mutagenesis in human genes causing genetic disease. Hum. Genet. 1991, 87, 409–415. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987. [Google Scholar] [CrossRef] [Green Version]
- McCullum, E.O.; Williams, B.A.R.; Zhang, J.; Chaput, J.C. Random Mutagenesis by Error-Prone PCR. Methods Mol Biol. 2010, 634, 103–109. [Google Scholar]
- Zubradt, M.; Gupta, P.; Persad, S.; Lambowitz, A.M.; Weissman, J.S.; Rouskin, S. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 2017, 14, 75–82. [Google Scholar] [CrossRef]
- Filler, A.G.; Lever, A.M.L. Effects of Cation Substitutions on Reverse Transcriptase and on Human Immunodeficiency Virus Production. AIDS Res. Hum. Retrovir. 1997, 13, 291–299. [Google Scholar] [CrossRef]
- Bolton, E.C.; Mildvan, A.S.; Boeke, J.D. Inhibition of Reverse Transcription In Vivo by Elevated Manganese Ion Concentration. Mol. Cell 2002, 9, 879–889. [Google Scholar] [CrossRef]
- Achuthan, V.; DeStefano, J.J. Alternative divalent cations (Zn2+, Co2+, and Mn2+) are not mutagenic at conditions optimal for HIV-1 reverse transcriptase activity. BMC Biochem. 2015, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Yarrington, R.M.; Chen, J.; Bolton, E.C.; Boeke, J.D. Mn2+ Suppressor Mutations and Biochemical Communication between Ty1 Reverse Transcriptase and RNase H Domains. J. Virol. 2007, 81, 9004–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. “FastQC.” Babraham Institute. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 February 2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Juhling, F.; Morl, M.; Hartmann, R.K.; Sprinzl, M.; Stadler, P.F.; Putz, J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009, 37, D159–D162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref_Seg | Mod | Pos | Ref_Base | Cov | Pre_Base | Mism_Rate | A | G | T | C | N | a | g | t | c | n | Single_Jump_Rate_Direct | Single_Jump Rate_Delayed | Double_Jump_Rate | Arrest_Rate |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tdbR00000369|Saccharomyces_cerevisiae|4932|Arg|ACG | m1A | 58 | A | 167 | G | 0.94012 | 10 | 39 | 24 | 7 | 0 | 0 | 58 | 25 | 4 | 0 | 0.00000 | 0.01754 | 0.01754 | 0.23661 |
| tdbR00000370|Saccharomyces_cerevisiae|4932|Arg|TCT | m1A | 57 | A | 25 | C | 0.52000 | 12 | 0 | 0 | 6 | 1 | 0 | 0 | 0 | 6 | 0 | 0.00000 | 0.00000 | 0.00000 | 0.03846 |
| tdbR00000300|Saccharomyces_cerevisiae|4932|Asn|GTT | m1A | 59 | A | 553 | C | 0.93671 | 35 | 4 | 7 | 229 | 1 | 0 | 5 | 11 | 261 | 0 | 0.00318 | 0.08493 | 0.40552 | 0.09519 |
| tdbR00000021|Saccharomyces_cerevisiae|4932|Cys|GCA | m1A | 57 | A | 562 | T | 0.94128 | 33 | 12 | 205 | 17 | 2 | 0 | 18 | 266 | 9 | 0 | 0.00000 | 0.00000 | 0.00858 | 0.09048 |
| tdbR00000170|Saccharomyces_cerevisiae|4932|Ile|AAT | m1A | 59 | A | 1177 | T | 0.96517 | 41 | 15 | 447 | 74 | 4 | 0 | 16 | 540 | 40 | 0 | 0.00168 | 0.00168 | 0.00587 | 0.23181 |
| tdbM00000006|Saccharomyces_cerevisiae|4932|Ile|TAT | m1A | 58 | A | 447 | T | 0.85682 | 64 | 15 | 138 | 23 | 1 | 0 | 25 | 161 | 20 | 0 | 0.00000 | 0.00665 | 0.00000 | 0.46437 |
| tdbR00000251|Saccharomyces_cerevisiae|4932|Leu|TAA | m1A | 69 | A | 571 | A | 0.35902 | 366 | 16 | 58 | 57 | 1 | 0 | 13 | 37 | 23 | 0 | 0.00520 | 0.00000 | 0.00173 | 0.43098 |
| tdbR00000250|Saccharomyces_cerevisiae|4932|Leu|TAG | m1A | 67 | A | 544 | A | 0.55699 | 241 | 17 | 96 | 41 | 2 | 0 | 22 | 87 | 38 | 0 | 0.01961 | 0.00000 | 0.00713 | 0.26988 |
| tdbR00000192|Saccharomyces_cerevisiae|4932|Lys|CTT | m1A | 58 | A | 604 | G | 0.72682 | 165 | 90 | 85 | 12 | 2 | 0 | 127 | 114 | 9 | 0 | 0.03343 | 0.00304 | 0.04407 | 0.17955 |
| tdbR00000193|Saccharomyces_cerevisiae|4932|Lys|TTT | m1A | 58 | A | 257 | G | 0.86381 | 35 | 36 | 53 | 9 | 1 | 0 | 46 | 69 | 8 | 0 | 0.07190 | 0.00000 | 0.06863 | 0.19895 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kristen, M.; Plehn, J.; Marchand, V.; Friedland, K.; Motorin, Y.; Helm, M.; Werner, S. Manganese Ions Individually Alter the Reverse Transcription Signature of Modified Ribonucleosides. Genes 2020, 11, 950. https://doi.org/10.3390/genes11080950
Kristen M, Plehn J, Marchand V, Friedland K, Motorin Y, Helm M, Werner S. Manganese Ions Individually Alter the Reverse Transcription Signature of Modified Ribonucleosides. Genes. 2020; 11(8):950. https://doi.org/10.3390/genes11080950
Chicago/Turabian StyleKristen, Marco, Johanna Plehn, Virginie Marchand, Kristina Friedland, Yuri Motorin, Mark Helm, and Stephan Werner. 2020. "Manganese Ions Individually Alter the Reverse Transcription Signature of Modified Ribonucleosides" Genes 11, no. 8: 950. https://doi.org/10.3390/genes11080950
APA StyleKristen, M., Plehn, J., Marchand, V., Friedland, K., Motorin, Y., Helm, M., & Werner, S. (2020). Manganese Ions Individually Alter the Reverse Transcription Signature of Modified Ribonucleosides. Genes, 11(8), 950. https://doi.org/10.3390/genes11080950