Basal Levels of CD18 Antigen Presenting Cells in Cow Milk Associate with Copy Number Variation of Fc Gamma Receptors

 ,
,  ,
,
Abstract
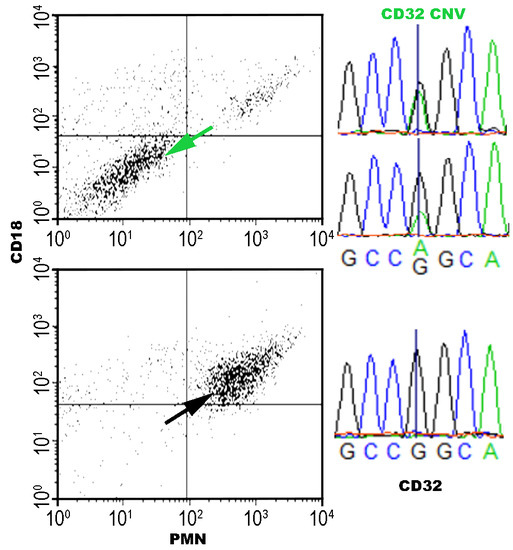
:
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Scheme
2.2. Antibodies, Conjugates, and Differential SCC
2.3. The Dataset, Haplotype Phasing, and Trait-Association Analysis
2.4. FCGR2 Exon 3 Sequencing and Peak Height Analysis
2.5. Sequence Read Archive (SRA) Search
2.6. Deep Sequencing and Analysis of Bovine Genomes
2.7. Statistical Analysis
3. Results
3.1. Genome-Wide Association Study (GWAS) of Immunological Traits
3.2. Analysis of Candidate Genes
3.3. CD32 vs. CD18 Gene Expression
3.4. Computerized Cloning of FCGR Genes of an Influential Israeli Holstein Sire
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Halasa, T.; Huijps, K.; Osteras, O.; Hogeveen, H. Economic effects of bovine mastitis and mastitis management: A review. Vet. Q. 2007, 29, 18–31. [Google Scholar] [CrossRef]
- Abebe, R.; Hatiya, H.; Abera, M.; Megersa, B.; Asmare, K. Bovine mastitis: Prevalence, risk factors and isolation of Staphylococcus aureus in dairy herds at Hawassa milk shed, South Ethiopia. BMC Vet. Res. 2016, 12, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, G.; Blum, S.E.; Krifuks, O.; Edery, N.; Merin, U. Correlation between Milk Bacteriology, Cytology and Mammary Tissue Histology in Cows: Cure from the Pathogen or Recovery from the Inflammation. Pathogens 2020, 9, 364. [Google Scholar] [CrossRef] [PubMed]
- Leitner, G.; Shoshani, E.; Krifucks, O.; Chaffer, M.; Saran, A. Milk leucocyte population patterns in bovine udder infection of different aetiology. J. Vet. Med. B 2000, 47, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Paape, M.J.; Shafer-Weaver, K.; Capuco, A.V.; Van Oostveldt, K.; Burvenich, C. Immune Surveillance of Mammary Tissue by Phagocytic Cells. In Biology of the Mammary Gland; Springer US: Boston, MA, USA, 2002; pp. 259–277. [Google Scholar]
- Seroussi, E.; Blum, S.E.; Krifucks, O.; Lavon, Y.; Leitner, G. Application of pancreatic phospholipase A2 for treatment of bovine mastitis. PLoS ONE 2018, 13, e0203132. [Google Scholar] [CrossRef]
- Oliver, S.P.; Gonzalez, R.N.; Hogan, J.S.; Jayarao, B.M.; Owens, W.E. Microbiological Procedures for the Diagnosis of Bovine Udder Infection and Determination of Milk Quality, 4th ed.; The National Mastitis Council, Inc.: Verona, WI, USA, 2004. [Google Scholar]
- Leitner, G.; Eligulashvily, R.; Krifucks, O.; Perl, S.; Saran, A. Immune cell differentiation in mammary gland tissues and milk of cows chronically infected with Staphylococcus aureus. J. Vet. Med. Ser. B 2003, 50, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [Green Version]
- NCBI. Sequence Read Archive Nucleotide BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&PROG_DEFAULTS=on&BLAST_SPEC=SRA (accessed on 30 June 2020).
- Bonfield, J.K.; Whitwham, A. Gap5—Editing the billion fragment sequence assembly. Bioinformatics 2010, 26, 1699–1703. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R-Core-Team. R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 30 June 2020).
- Fanciulli, M.; Vyse, T.J.; Aitman, T.J. Copy number variation of Fc γ receptor genes and disease predisposition. Cytogenet. Genome Res. 2008, 123, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Mott, R.; Flint, J. Dissecting quantitative traits in mice. Annu. Rev. Genom. Hum. Genet. 2013, 14, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.S.; Guldbrandtsen, B.; Buitenhuis, A.J.; Thomsen, B.; Bendixen, C. Detection of quantitative trait loci in Danish Holstein cattle affecting clinical mastitis, somatic cell score, udder conformation traits, and assessment of associated effects on milk yield. J. Dairy Sci. 2008, 91, 4028–4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobbo, T.; Penasa, M.; Cassandro, M. Short communication: Genetic aspects of milk differential somatic cell count in Holstein cows: A preliminary analysis. J. Dairy Sci. 2019, 102, 4275–4279. [Google Scholar] [CrossRef]
- Montojo, J.; Zuberi, K.; Rodriguez, H.; Bader, G.D.; Morris, Q. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Res 2014, 3, 153. [Google Scholar] [CrossRef]
- Breunis, W.B.; van Mirre, E.; Geissler, J.; Laddach, N.; Wolbink, G.; van der Schoot, E.; de Haas, M.; de Boer, M.; Roos, D.; Kuijpers, T.W. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum. Mutat. 2009, 30, E640–E650. [Google Scholar] [CrossRef]
- Koks, S.; Reimann, E.; Lilleoja, R.; Lattekivi, F.; Salumets, A.; Reemann, P.; Jaakma, U. Sequencing and annotated analysis of full genome of Holstein breed bull. Mamm. Genome 2014, 25, 363–373. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Hou, Y.L.; Schroeder, S.G.; Alkan, C.; Cardone, M.F.; Matukumalli, L.K.; Song, J.Z.; Schnabe, R.D.; Ventura, M.; Taylor, J.F.; et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012, 22, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Seroussi, E.; Klompus, S.; Silanikove, M.; Krifucks, O.; Shapiro, F.; Gertler, A.; Leitner, G. Nonbactericidal secreted phospholipase A2s are potential anti-inflammatory factors in the mammary gland. Immunogenetics 2013, 65, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Shirak, A.; Seroussi, U.; Gootwine, E.; Seroussi, E. Sequence motifs capable of forming DNA stem-loop structures act as a replication diode. FEBS Open Bio 2017, 7, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Golik, M.; Cohen-Zinder, M.; Loor, J.J.; Drackley, J.K.; Band, M.R.; Lewin, H.A.; Weller, J.I.; Ron, M.; Seroussi, E. Accelerated expansion of group IID-like phospholipase A2 genes in Bos taurus. Genomics 2006, 87, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainard, P.; Foucras, G.; Boichard, D.; Rupp, R. Invited review: Low milk somatic cell count and susceptibility to mastitis. J. Dairy Sci. 2018, 101, 6703–6714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, A.L.; Leitner, G.; Merin, U. Milk quality and udder health. Test methods and standards. In Encyclopedia of Dairy Sciences, 2nd ed.; Fuquay, J.W., Fox, P.F., McSweeney, P.L.H., Eds.; Academic Press: San Diego, CA, USA, 2011; pp. 894–901. [Google Scholar]
- Leitner, G.; Merin, U.; Krifucks, O.; Blum, S.; Rivas, A.L.; Silanikove, N. Effects of intra-mammary bacterial infection with coagulase negative Staphylococci and stage of lactation on shedding of epithelial cells and infiltration of leukocytes into milk: Comparison among cows, goats and sheep. Vet. Immunol. Immunopathol. 2012, 147, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.; Burvenich, C.; Guidry, A.J.; Massart-Leen, A. Adhesion receptor CD11b/CD18 contributes to neutrophil diapedesis across the bovine blood-milk barrier. Vet. Immunol. Immunopathol. 2000, 73, 255–265. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | HAPLOTYPE | F 2 | β | STAT | P | EMP1 | EMP2 |
|---|---|---|---|---|---|---|---|
| 1 | GGTGAGAACGAAGGG | 0.08 | −1.17 × 103 | 0.46 | 0.499 | 0.504 | 1.00 |
| 2 | AAAGGAGAAGGGAAG | 0.07 | −2.17 × 103 | 1.66 | 0.200 | 0.199 | 1.00 |
| 3 | AGTGAGAAAAGAAGA | 0.06 | −2.25 × 103 | 1.17 | 0.281 | 0.282 | 1.00 |
| 4 | GGTGGGGGAGGAAAG | 0.05 | 1.16 × 104 | 36.00 | 2.54 × 10−8 | 5.00 × 10−6 | 1.00 × 10−5 |
| 5 | GGTGAGAGCAAGAAG | 0.04 | 2.11 × 103 | 0.58 | 0.447 | 0.454 | 1.00 |
| 6 | GGTGAGAAAGGAAGG | 0.04 | −593 | 0.08 | 0.780 | 0.781 | 1.00 |
| 7 | GGTAAGAAAGAAAAG | 0.03 | −4.07 × 103 | 1.68 | 0.198 | 0.198 | 1.00 |
| 8 | GGTGAGGGCGAGAAG | 0.03 | −375 | 0.03 | 0.871 | 0.872 | 1.00 |
| 9 | GGTAGAAGAAAGAGG | 0.03 | 4.91 × 103 | 2.06 | 0.154 | 0.150 | 0.98 |
| 10 | AGTGAGAAAGGGAAG | 0.03 | −550 | 0.04 | 0.849 | 0.850 | 1.00 |
| 11 | AAAAGAAGCGGAAGG | 0.03 | 1.97 × 103 | 0.39 | 0.534 | 0.541 | 1.00 |
| 12 | AGTAGGGGAAGAAAG | 0.03 | −2.25 × 103 | 0.34 | 0.559 | 0.572 | 1.00 |
| 13 | AGTAGAAGCGGAAGG | 0.03 | −1.99 × 103 | 0.33 | 0.564 | 0.576 | 1.00 |
| 14 | GGTGGAAAAGGAAGG | 0.03 | 3.50 × 103 | 0.63 | 0.429 | 0.449 | 1.00 |
| 15 | GGTGAGAACAAGGGG | 0.02 | −3.13 × 103 | 1.33 | 0.251 | 0.253 | 1.00 |
| 16 | AGTGGAAAAGAAAGG | 0.02 | 5.67 × 103 | 3.83 | 0.053 | 0.049 | 0.70 |
| 17 | GGTGAGAAAAGAAGA | 0.02 | −4.01 × 103 | 1.97 | 0.163 | 0.162 | 0.99 |
| 18 | AGAGAAGACGAAAAG | 0.02 | −428 | 0.02 | 0.902 | 0.905 | 1.00 |
| 19 | AGAGAAGACGAAGGG | 0.01 | −1.53 × 103 | 0.20 | 0.658 | 0.668 | 1.00 |
| 20 | GAAAAGAACAAGGGG | 0.01 | −864 | 0.03 | 0.873 | 0.898 | 1.00 |
| 21 | GGTGAGAGAAAGGAG | 0.01 | 3.57 × 103 | 0.87 | 0.353 | 0.365 | 1.00 |
| 22 | GGTAGAAAAGGAAAG | 0.01 | 480 | 0.01 | 0.929 | 0.944 | 1.00 |
| 23 | GGTGAGAACGAAAAG | 0.01 | −1.45 × 103 | 0.14 | 0.706 | 0.716 | 1.00 |
| 24 | GGTGAGAAAGGAGGG | 0.01 | 3.44 × 103 | 1.00 | 0.319 | 0.325 | 1.00 |
| Milk | Blood | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Reads | RPKM | Number of Reads | RPKM | |||||||||||||||
| # 1 | SRA | Total | 1 | 4 | 7 | 9 | Sum 2 | CD32 | CD18 | R 3 | SRA | Total | 1 | 4 | 7 | 9 | CD32 | CD18 |
| 1 | 394 | 27,331,010 | 1 | 17 | 6 | 176 | 200 | 27.4 | 201.2 | 270 | 8,584,110 | 0 | 5 | 5 | 104 | 36.4 | 378.6 | |
| 2 | 376 | 19,981,694 | 5 | 7 | 7 | 205 | 225 | 31.3 | 320.6 | 226 | 21,522,312 | 0 | 2 | 2 | 173 | 5.8 | 251.2 | |
| 3 | 254 | 18,609,806 | 0 | 13 | 0 | 219 | 232 | 21.8 | 367.7 | 250 | 1,177,238 | 0 | 0 | 0 | 16 | 0.0 | 424.7 | |
| 4 | 367 | 16,301,126 | 3 | 2 | 10 | 220 | 235 | 28.8 | 421.8 | 218 | 22,950,370 | 0 | 13 | 17 | 192 | 40.8 | 261.4 | |
| 5 | 368 | 20,876,820 | 0 | 19 | 7 | 209 | 235 | 38.9 | 312.8 | 170 | 14,416,000 | 0 | 17 | 16 | 164 | 71.5 | 355.5 | |
| 6 | 340 | 21,342,290 | 10 | 54 | 0 | 173 | 237 | 93.7 | 253.3 | 235 | 13,338,032 | 0 | 26 | 0 | 144 | 60.9 | 337.4 | |
| 7 | 341 | 25,687,198 | 4 | 42 | 0 | 203 | 249 | 56.0 | 247.0 | 236 | 16,838,534 | 1 | 26 | 0 | 406 | 50.1 | 753.5 | |
| 8 | 210 | 14,517,104 | 2 | 6 | 9 | 235 | 252 | 36.6 | 505.9 | 257 | 9,046,998 | 0 | 0 | 7 | 103 | 27.6 | 355.8 | |
| 9 | 361 | 23,695,512 | 0 | 22 | 9 | 247 | 279 | 42.2 | 325.7 | 207 | 13,028,116 | 0 | 15 | 8 | 274 | 55.2 | 657.2 | |
| 10 | 232 | 17,678,968 | 0 | 0 | 3 | 306 | 309 | 5.3 | 540.9 | −0.55 | 168 | 23,333,558 | 0 | 2 | 34 | 122 | 48.2 | 163.4 |
| 11 | 188 | 40,398,796 | 3 | 42 | 8 | 277 | 330 | 41.0 | 214.3 | −0.65 | 214 | 23,555,514 | 1 | 26 | 20 | 223 | 63.7 | 295.8 |
| 12 | 345 | 26,493,242 | 2 | 40 | 0 | 299 | 341 | 49.5 | 352.7 | −0.67 | 193 | 23,755,222 | 0 | 51 | 0 | 307 | 67.1 | 403.9 |
| 13 | 318 | 41,897,140 | 0 | 177 | 103 | 68 | 348 | 208.8 | 50.7 | −0.81 | 229 | 6,410,488 | 0 | 9 | 2 | 59 | 53.6 | 287.6 |
| 14 | 321 | 45,013,510 | 15 | 32 | 32 | 362 | 441 | 54.8 | 251.3 | −0.79 | 230 | 21,228,336 | 1 | 19 | 19 | 303 | 504.9 | 446.0 |
| 15 | 365 | 18,179,706 | 0 | 7 | 3 | 439 | 449 | 17.2 | 754.6 | −0.73 | 258 | 11,983,986 | 0 | 9 | 4 | 157 | 33.9 | 409.4 |
| 16 | 316 | 30,449,572 | 0 | 24 | 15 | 410 | 449 | 40.0 | 420.8 | −0.72 | 261 | 3,872,402 | 0 | 7 | 1 | 61 | 64.6 | 492.3 |
| 17 | 243 | 28,918,832 | 8 | 22 | 12 | 443 | 485 | 45.4 | 478.7 | −0.74 | 217 | 18,766,692 | 3 | 19 | 11 | 214 | 55.0 | 356.3 |
| 18 | 349 | 30,480,908 | 10 | 88 | 0 | 541 | 639 | 100.5 | 554.7 | −0.61 | 273 | 8,150,358 | 0 | 12 | 0 | 119 | 46.0 | 456.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seroussi, E.; Blum, S.E.; Krifucks, O.; Shirak, A.; Jacoby, S.; Leitner, G. Basal Levels of CD18 Antigen Presenting Cells in Cow Milk Associate with Copy Number Variation of Fc Gamma Receptors. Genes 2020, 11, 952. https://doi.org/10.3390/genes11080952
Seroussi E, Blum SE, Krifucks O, Shirak A, Jacoby S, Leitner G. Basal Levels of CD18 Antigen Presenting Cells in Cow Milk Associate with Copy Number Variation of Fc Gamma Receptors. Genes. 2020; 11(8):952. https://doi.org/10.3390/genes11080952
Chicago/Turabian StyleSeroussi, Eyal, Shlomo E. Blum, Oleg Krifucks, Andrey Shirak, Shamay Jacoby, and Gabriel Leitner. 2020. "Basal Levels of CD18 Antigen Presenting Cells in Cow Milk Associate with Copy Number Variation of Fc Gamma Receptors" Genes 11, no. 8: 952. https://doi.org/10.3390/genes11080952