Genome-Wide DNA Methylation Signatures of Sea Cucumber Apostichopus japonicus during Environmental Induced Aestivation

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Total RNA Extraction and Quantitative RT-PCR
2.3. Genomic DNA Extraction and Whole-Genome Bisulfite Sequencing
2.4. Identification of Differentially Methylated Regions
2.5. Gene Ontology and Pathway Enrichment of DMRs
3. Results
3.1. Expression of DNA Methylation Related Enzymes during Aestivation
3.2. DNA Methylation of Sea Cucumber during Aestivation
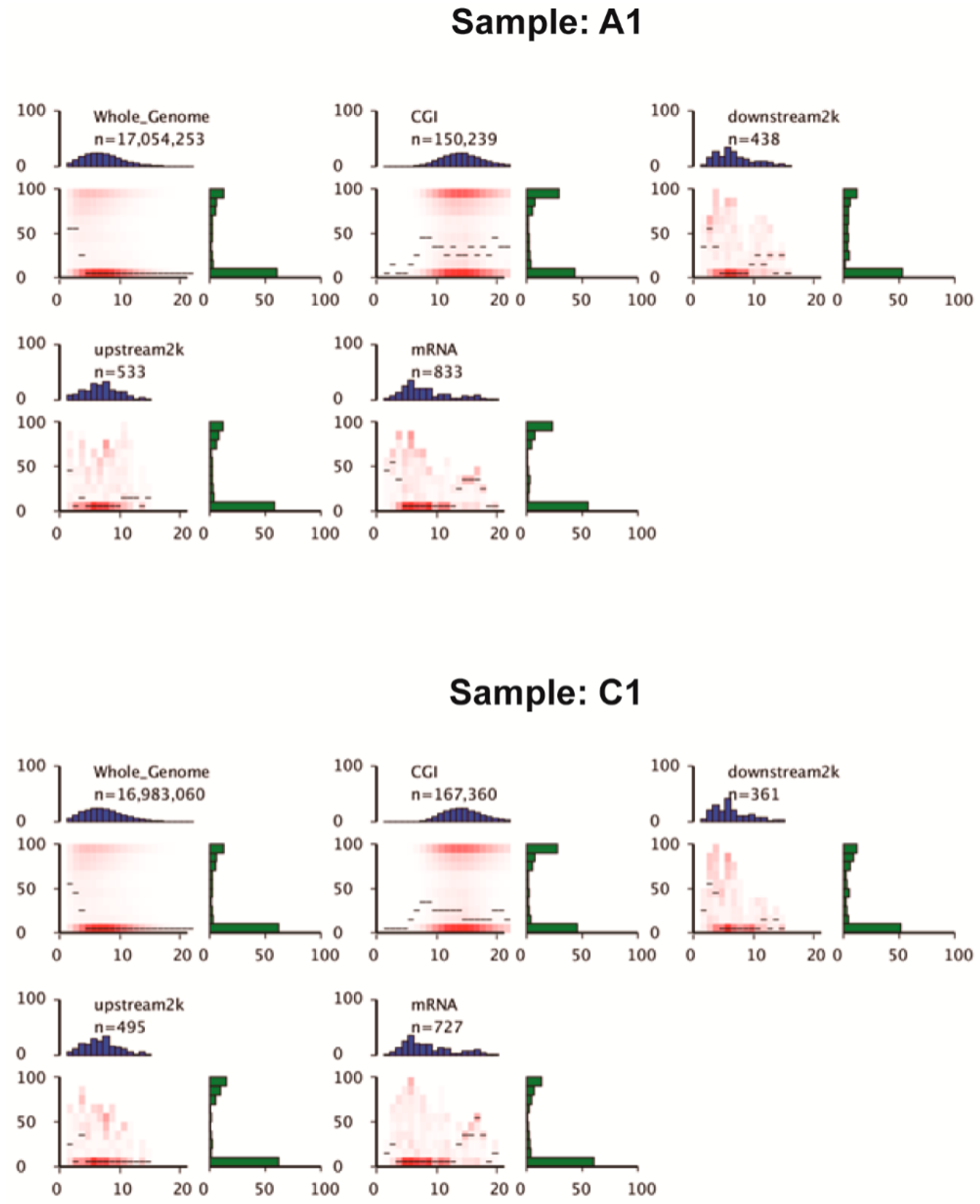
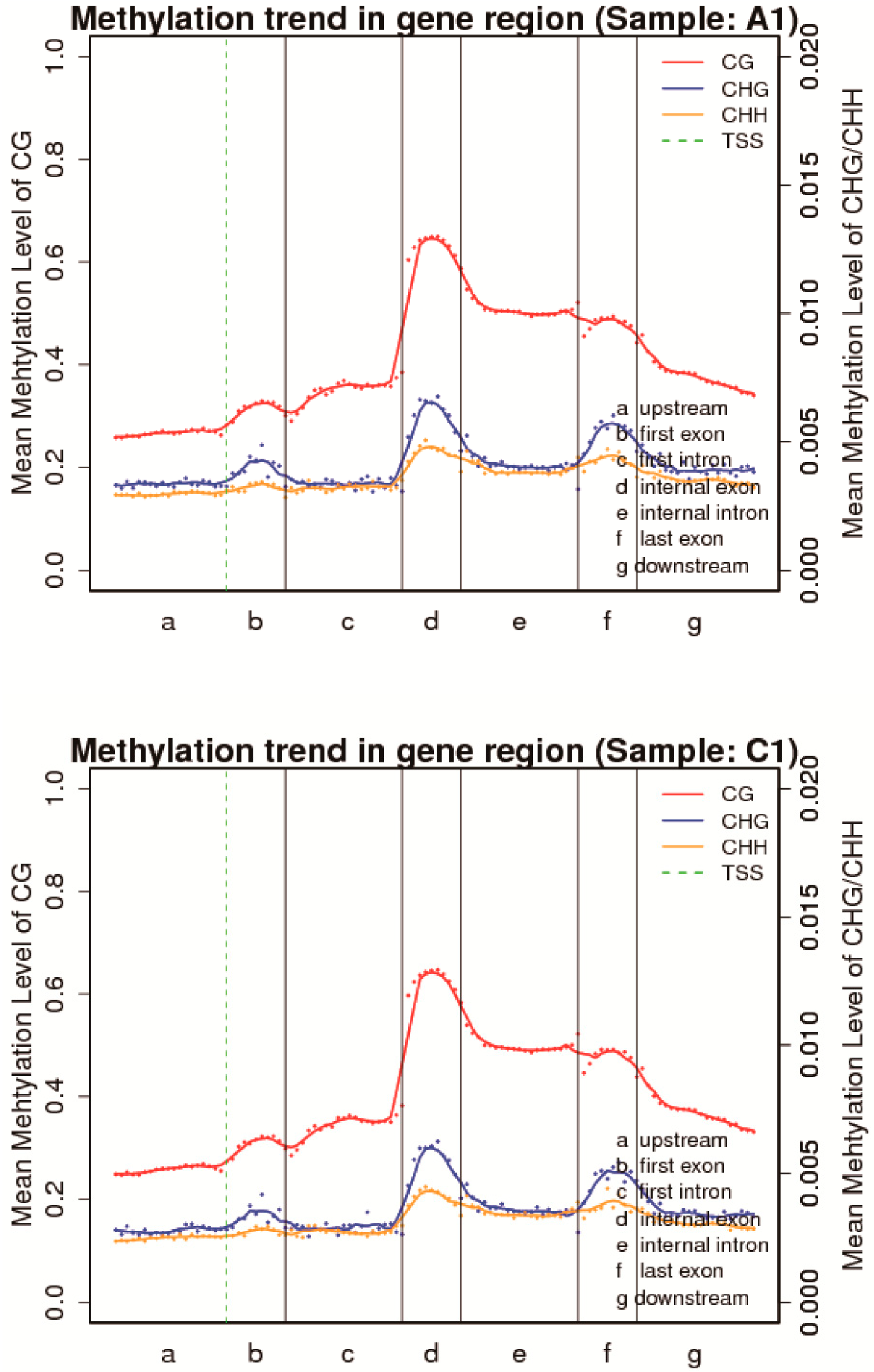
3.3. Proportion of Methylation Contexts and mCs Distribution across Genomic Features
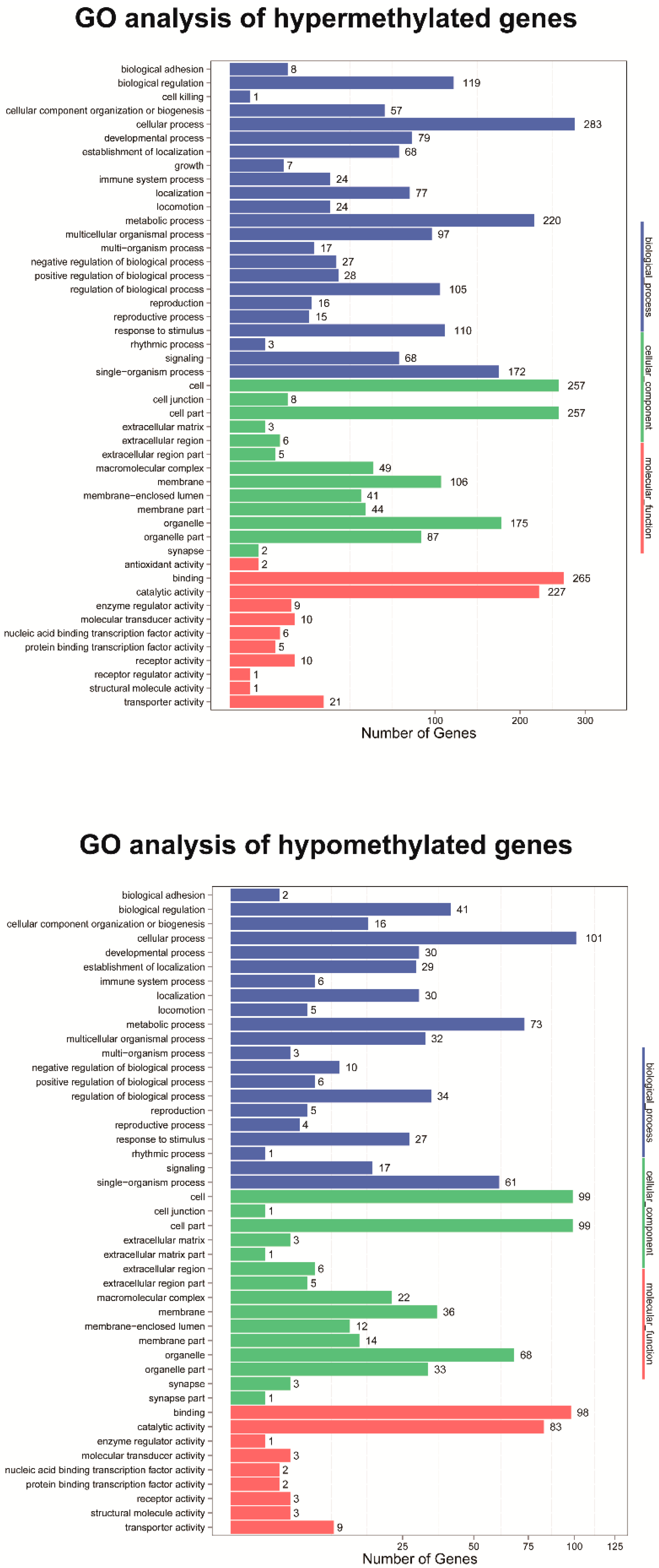
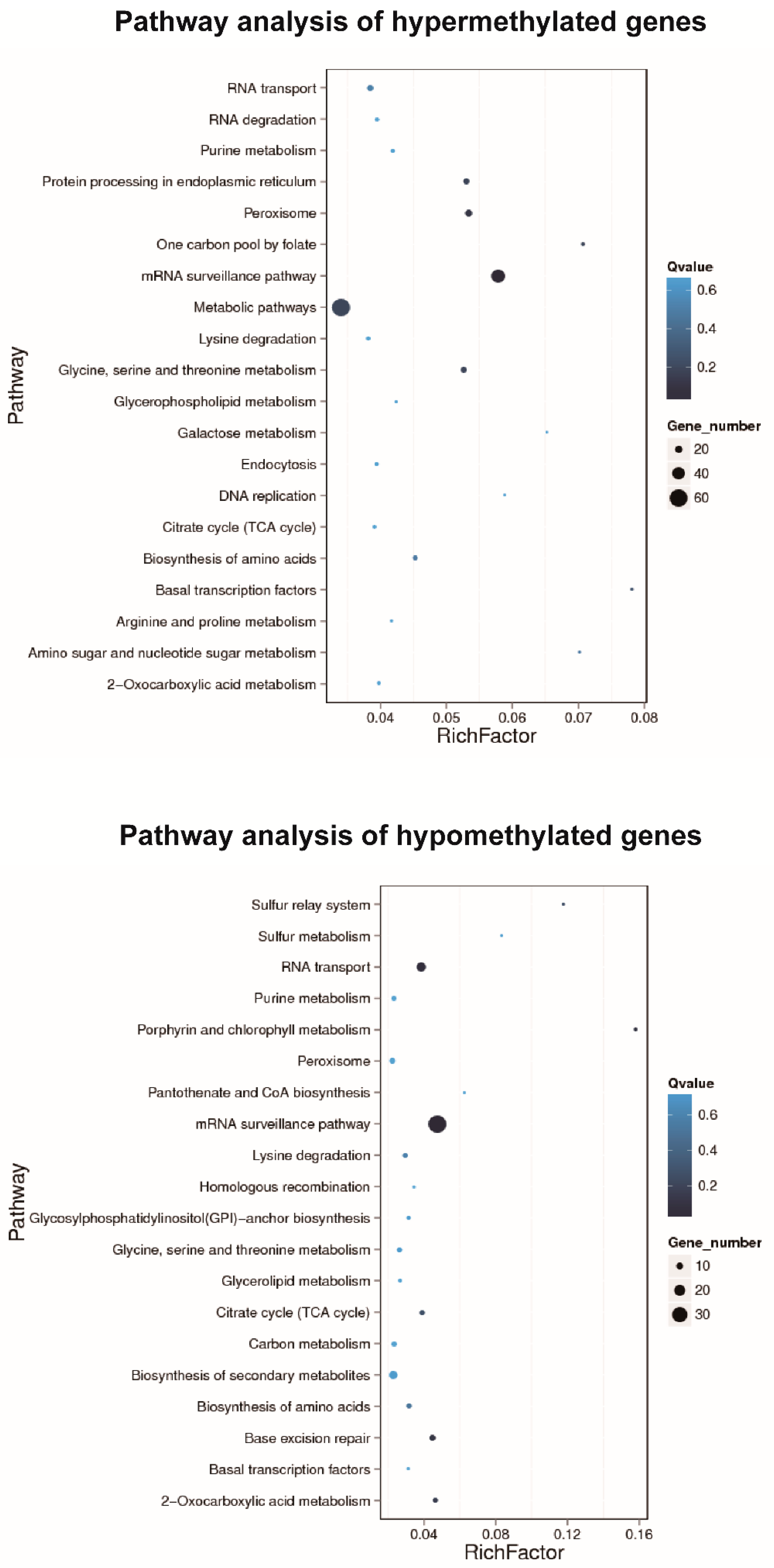
3.4. Identification and Enrichment Analysis of Differential Methylated Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Price, T.D.; Qvarnström, A.; Irwin, D.E. The role of phenotypic plasticity in driving genetic evolution. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 1433–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldmaier, G.; Ortmann, S.; Elvert, R. Natural hypometabolism during hibernation and daily torpor in mammals. Respir. Physiol. Neurobiol. 2004, 141, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Storey, K.B.; Storey, J.M. Aestivation: Signaling and hypometabolism. J. Exp. Biol. 2012, 215, 1425–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renfree, M.B.; Shaw, G. Diapause. Annu. Rev. Physiol. 2000, 62, 353–375. [Google Scholar] [CrossRef]
- van Breukelen, F.; Martin, S.L. The hibernation continuum: Physiological and molecular aspects of metabolic plasticity in mammals. Physiology 2015, 30, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Canale, C.I.; Henry, P.-Y. Adaptive phenotypic plasticity and resilience of vertebrates to increasing climatic unpredictability. Clim. Res. 2010, 43, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Winterhalter, W.E.; Mousseau, T.A. Patterns of phenotypic and genetic variation for the plasticity of diapause incidence. Evol. Int. J. Org. Evol. 2007, 61, 1520–1531. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation through repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Kronholm, I.; Collins, S. Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 2016, 25, 1856–1868. [Google Scholar] [CrossRef]
- Wilschut, R.A.; Oplaat, C.; Snoek, L.B.; Kirschner, J.; Verhoeven, K.J. Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Mol. Ecol. 2016, 25, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.; Storey, K.B. Mammalian hibernation: Differential gene expression and novel application of epigenetic controls. Int. J. Dev. Biol. 2009, 53, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Storey, K.B. Regulation of hypometabolism: Insights into epigenetic controls. J. Exp. Biol. 2015, 218, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessier, S.N.; Luu, B.E.; Smith, J.C.; Storey, K.B. The role of global histone post-translational modifications during mammalian hibernation. Cryobiology 2017, 75, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Jeremias, G.; Barbosa, J.; Marques, S.M.; Asselman, J.; Gonçalves, F.J.; Pereira, J.L. Synthesizing the role of epigenetics in the response and adaptation of species to climate change in freshwater ecosystems. Mol. Ecol. 2018, 27, 2790–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, S.B.; Gavery, M.R. Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front. Physiol. 2012, 2, 116. [Google Scholar] [CrossRef] [Green Version]
- Gavery, M.R.; Roberts, S.B. DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genom. 2010, 11, 483. [Google Scholar] [CrossRef] [Green Version]
- Putnam, H.M.; Davidson, J.M.; Gates, R.D. Ocean acidification influences host DNA methylation and phenotypic plasticity in environmentally susceptible corals. Evol. Appl. 2016, 9, 1165–1178. [Google Scholar] [CrossRef]
- Storey, K.B.; Storey, J.M. Metabolic Regulation and Gene Expression during Aestivation. In Aestivation; Springer: Berlin/Heidelberg, Germany, 2010; pp. 25–45. [Google Scholar]
- Wang, F.; Yang, H.; Gao, F.; Liu, G. Effects of acute temperature or salinity stress on the immune response in sea cucumber, Apostichopus japonicus. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2008, 151, 491–498. [Google Scholar] [CrossRef]
- Yang, H.; Hamel, J.-F.; Mercier, A. The Sea Cucumber Apostichopus Japonicus: History, Biology and Aquaculture; Academic Press: London, UK, 2015; Volume 39. [Google Scholar]
- Gao, F.; Yang, H.; Xu, Q.; Wang, F.; Liu, G.; German, D.P. Phenotypic plasticity of gut structure and function during periods of inactivity in Apostichopus japonicus. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2008, 150, 255–262. [Google Scholar] [CrossRef]
- Zhu, A.; Chen, M.; Zhang, X.; Storey, K.B. Gene structure, expression, and DNA methylation characteristics of sea cucumber cyclin B gene during aestivation. Gene 2016, 594, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, R.; Xun, X.; Wang, J.; Bao, L.; Thimmappa, R.; Ding, J.; Jiang, J.; Zhang, L.; Li, T. Sea cucumber genome provides insights into saponin biosynthesis and aestivation regulation. Cell Discov. 2018, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yang, H.; Xu, Q.; Wang, F.; Liu, G. Effect of water temperature on digestive enzyme activity and gut mass in sea cucumber Apostichopus japonicus (Selenka), with special reference to aestivation. Chin. J. Oceanol. Limnol. 2009, 27, 714. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, H.; Storey, K.B.; Chen, M. RNA-seq dependent transcriptional analysis unveils gene expression profile in the intestine of sea cucumber Apostichopus japonicus during aestivation. Comp. Biochem. Physiol. Part D Genom. Proteom. 2014, 10, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, M.; Wang, T.; Sun, L.; Xu, D.; Yang, H. Selection of reference genes for qRT-PCR analysis of gene expression in sea cucumber Apostichopus japonicus during aestivation. Chin. J. Oceanol. Limnol. 2014, 32, 1248–1256. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, L.; Yuan, J.; Sun, Y.; Gao, Y.; Zhang, L.; Li, S.; Dai, H.; Hamel, J.-F.; Liu, C. The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLoS Biol. 2017, 15, e2003790. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y. Single base–resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010, 8, e1000533. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Q.; Lian, J.; Li, L.; Jin, L.; Cai, H.; Xu, F.; Qi, H.; Zhang, L.; Wu, F. Genome-wide and single-base resolution DNA methylomes of the Pacific oyster Crassostrea gigas provide insight into the evolution of invertebrate CpG methylation. BMC Genom. 2014, 15, 1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, N.J.; Lonhienne, T.; Franklin, C.E.; Harper, G.S.; Lehnert, S. Epigenetic silencers are enriched in dormant desert frog muscle. J. Comp. Physiol. B 2008, 178, 729–734. [Google Scholar] [CrossRef]
- Krivoruchko, A.; Storey, K.B. Epigenetics in anoxia tolerance: A role for histone deacetylases. Mol. Cell. Biochem. 2010, 342, 151–161. [Google Scholar] [CrossRef]
- Seibel, B.A.; Häfker, N.S.; Trübenbach, K.; Zhang, J.; Tessier, S.N.; Pörtner, H.-O.; Rosa, R.; Storey, K.B. Metabolic suppression during protracted exposure to hypoxia in the jumbo squid, Dosidicus gigas, living in an oxygen minimum zone. J. Exp. Biol. 2014, 217, 2555–2568. [Google Scholar] [CrossRef] [Green Version]
- Wijenayake, S.; Hawkins, L.J.; Storey, K.B. Dynamic regulation of six histone H3 lysine (K) methyltransferases in response to prolonged anoxia exposure in a freshwater turtle. Gene 2018, 649, 50–57. [Google Scholar] [CrossRef]
- Wang, T.; Yang, H.; Zhao, H.; Chen, M.; Wang, B. Transcriptional changes in epigenetic modifiers associated with gene silencing in the intestine of the sea cucumber, Apostichopus japonicus (Selenka), during aestivation. Chin. J. Oceanol. Limnol. 2011, 29, 1267. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, M.; Su, L.; Wang, T.; Liu, S.; Yang, H. Molecular cloning and expression-profile analysis of sea cucumber DNA (Cytosine-5)-methyltransferase 1 and methyl-CpG binding domain type 2/3 genes during aestivation. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2013, 165, 26–35. [Google Scholar] [CrossRef]
- Alvarado, S.; Mak, T.; Liu, S.; Storey, K.B.; Szyf, M. Dynamic changes in global and gene-specific DNA methylation during hibernation in adult thirteen-lined ground squirrels, Ictidomys tridecemlineatus. J. Exp. Biol. 2015, 218, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Wijenayake, S.; Storey, K.B. The role of DNA methylation during anoxia tolerance in a freshwater turtle (Trachemys scripta elegans). J. Comp. Physiol. B 2016, 186, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Porter, J.; Sun, M.-A.; Xie, H.; Zhang, L. Objective and comprehensive evaluation of bisulfite short read mapping tools. Adv. Bioinform. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Cui, Y.; Gu, X. Genome-wide DNA methylation profiles changes associated with constant heat stress in pigs as measured by bisulfite sequencing. Sci. Rep. 2016, 6, 27507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Li, L.; Zhang, G. The association between DNA methylation and exon expression in the Pacific oyster Crassostrea gigas. PLoS ONE 2017, 12, e0185224. [Google Scholar] [CrossRef]
- Isken, O.; Maquat, L.E. Quality control of eukaryotic mRNA: Safeguarding cells from abnormal mRNA function. Genes Dev. 2007, 21, 1833–3856. [Google Scholar] [CrossRef] [Green Version]
- Jamar, N.H.; Kritsiligkou, P.; Grant, C.M. The non-stop decay mRNA surveillance pathway is required for oxidative stress tolerance. Nucleic Acids Res. 2017, 45, 6881–6893. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Burkhardt, D.H.; Rouskin, S.; Li, G.-W.; Weissman, J.S.; Gross, C.A. A stress response that monitors and regulates mRNA structure is central to cold shock adaptation. Mol. Cell 2018, 70, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Terzian, C.; Santamaria, P.; Pelisson, A.; Purd'homme, N.; Bucheton, A. Retroviruses in invertebrates: The gypsy retrotransposon is apparently an infectious retrovirus of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1994, 91, 1285–1289. [Google Scholar] [CrossRef] [Green Version]
- Whitelaw, E.; Martin, D.I. Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat. Genet. 2001, 27, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.D.; Miller, L.P.; Kaluziak, S.T.; Trussell, G.C.; Vollmer, S.V. Thermal stress and predation risk trigger distinct transcriptomic responses in the intertidal snail Nucella lapillus. Mol. Ecol. 2014, 23, 6104–6113. [Google Scholar] [CrossRef] [PubMed]
- Traylor-Knowles, N.; Rose, N.H.; Sheets, E.A.; Palumbi, S.R. Early transcriptional responses during heat stress in the coral Acropora hyacinthus. Biol. Bull. 2017, 232, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Menike, U.; Lee, Y.; Oh, C.; Wickramaarachchi, W.; Premachandra, H.; Park, S.C.; Lee, J.; De Zoysa, M. Oligo-microarray analysis and identification of stress-immune response genes from manila clam (Ruditapes philippinarum) exposure to heat and cold stresses. Mol. Biol. Rep. 2014, 41, 6457–6473. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wessler, S.R. Transposable elements and the evolution of eukaryotic genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 17600–17601. [Google Scholar] [CrossRef] [Green Version]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and diversity of transposable elements in vertebrate genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Mita, P.; Boeke, J.D. How retrotransposons shape genome regulation. Curr. Opin. Genet. Dev. 2016, 37, 90–100. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | Sequences |
|---|---|
| Dnmt1 | F: ATATGGCGTCCCACAGACTC |
| R: CACAGTGATGGTGCGATAGG | |
| Dnmt3a | F: CAGACGCGATCAAAGTCTCA |
| R: CCCCAATCAACAGGTCAAAT | |
| Dnmt3b | F: CGTCGTTACAGAGGGGAGAG |
| R: ACCACTTTCAGGGAGGGACT | |
| MBD2/3 | F: CTTTGACCTTAGCGCCTTTG |
| R: GTTCTGGGCGTGATTTGTTT | |
| MBD4 | F: ACCAGTCCGAATCAACCAAC |
| R: AACCAATCCTCGTCAGTTCC | |
| MBD5 | F: ACGGAGATGTCAAAGGGCAG |
| R: GCTGCCTCCAAGTTAGTCGT | |
| MBD6 | F: ATCGTCTCCTGCCTCCTCTT |
| R: GATGCTGCTGTGCTTCGATG | |
| TDG | F: CGTTATGCCTTCGTCAAGTG |
| R: ATTCACCGCCTTCAGTTCAT | |
| TET2 | F: ACCAGCAGAGGAAGGGAAAT |
| R: TGGAGGCATACGAGGCTACT |
| Sample ID | Clean Reads | Clean Data Size(bp) | Mapped Reads | Mapping Rate (%) | Uniquely Mapped Reads | Uniquely Mapping Rate (%) | Bisulfite Conversion Rate (%) |
|---|---|---|---|---|---|---|---|
| A1 | 235,240,966 | 29,405,120,750 | 146,748,774 | 62.38 | 120,561,967 | 51.25 | 99.83 |
| A2 | 221,903,100 | 27,737,887,500 | 138,682,431 | 62.50 | 114,085,412 | 51.41 | 99.82 |
| A3 | 252,389,374 | 31,548,671,750 | 158,828,900 | 62.93 | 130,699,039 | 51.78 | 99.83 |
| C1 | 214,743,374 | 26,842,921,750 | 134,186,828 | 62.49 | 109,061,960 | 50.79 | 99.87 |
| C2 | 223,167,750 | 27,895,968,750 | 141,594,447 | 63.45 | 114,184,023 | 51.17 | 99.90 |
| C3 | 240,490,792 | 30,061,349,000 | 151,604,875 | 63.04 | 124,088,420 | 51.60 | 99.89 |
| Sample | mCG | mCHG | mCHH | |
|---|---|---|---|---|
| A1 | mC number | 6,190,426 | 80,195 | 266,577 |
| Proportion (%) | 94.695 | 1.227 | 4.078 | |
| A2 | mC number | 5,984,027 | 77,057 | 256,981 |
| Proportion (%) | 94.713 | 1.22 | 4.067 | |
| A3 | mC number | 6,385,314 | 83,819 | 276,569 |
| Proportion (%) | 94.658 | 1.243 | 4.1 | |
| C1 | mC number | 6,017,278 | 76,797 | 253,064 |
| Proportion (%) | 94.803 | 1.21 | 3.987 | |
| C2 | mC number | 6,111,110 | 78,486 | 257,136 |
| Proportion (%) | 94.794 | 1.217 | 3.989 | |
| C3 | mC number | 5,752,504 | 71,494 | 235,918 |
| Proportion (%) | 94.927 | 1.18 | 3.893 |
| Group | Regions | C (%) | CG (%) | CHG (%) | CHH (%) |
|---|---|---|---|---|---|
| Aestivation Group 1 | genome | 3.564 | 28.113 | 0.331 | 0.293 |
| CDS | 7.079 | 43.595 | 0.478 | 0.487 | |
| intron | 2.649 | 20.231 | 0.341 | 0.295 | |
| mRNA | 3.463 | 26.968 | 0.342 | 0.291 | |
| Aestivation Group 2 | genome | 3.476 | 27.337 | 0.333 | 0.298 |
| CDS | 6.909 | 42.708 | 0.396 | 0.526 | |
| intron | 2.433 | 18.562 | 0.346 | 0.285 | |
| mRNA | 3.257 | 25.355 | 0.34 | 0.291 | |
| Aestivation Group 3 | genome | 3.429 | 27.199 | 0.329 | 0.294 |
| CDS | 7.022 | 44.404 | 0.374 | 0.472 | |
| intron | 2.467 | 18.98 | 0.355 | 0.293 | |
| mRNA | 3.316 | 25.896 | 0.338 | 0.295 | |
| Control Group 1 | genome | 3.521 | 27.1 | 0.281 | 0.25 |
| CDS | 7.479 | 46.443 | 0.408 | 0.491 | |
| intron | 2.602 | 19.658 | 0.298 | 0.256 | |
| mRNA | 3.414 | 26.121 | 0.289 | 0.258 | |
| Control Group 2 | genome | 3.329 | 24.957 | 0.248 | 0.217 |
| CDS | 6.966 | 43.446 | 0.331 | 0.455 | |
| intron | 2.476 | 18.712 | 0.248 | 0.219 | |
| mRNA | 3.255 | 24.56 | 0.246 | 0.232 | |
| Control Group 3 | genome | 3.529 | 27.534 | 0.269 | 0.235 |
| CDS | 7.192 | 43.552 | 0.432 | 0.627 | |
| intron | 2.62 | 19.591 | 0.288 | 0.232 | |
| mRNA | 3.342 | 25.364 | 0.268 | 0.258 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Zheng, Y.; Sun, L.; Chen, M. Genome-Wide DNA Methylation Signatures of Sea Cucumber Apostichopus japonicus during Environmental Induced Aestivation. Genes 2020, 11, 1020. https://doi.org/10.3390/genes11091020
Yang Y, Zheng Y, Sun L, Chen M. Genome-Wide DNA Methylation Signatures of Sea Cucumber Apostichopus japonicus during Environmental Induced Aestivation. Genes. 2020; 11(9):1020. https://doi.org/10.3390/genes11091020
Chicago/Turabian StyleYang, Yujia, Yingqiu Zheng, Lina Sun, and Muyan Chen. 2020. "Genome-Wide DNA Methylation Signatures of Sea Cucumber Apostichopus japonicus during Environmental Induced Aestivation" Genes 11, no. 9: 1020. https://doi.org/10.3390/genes11091020