The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
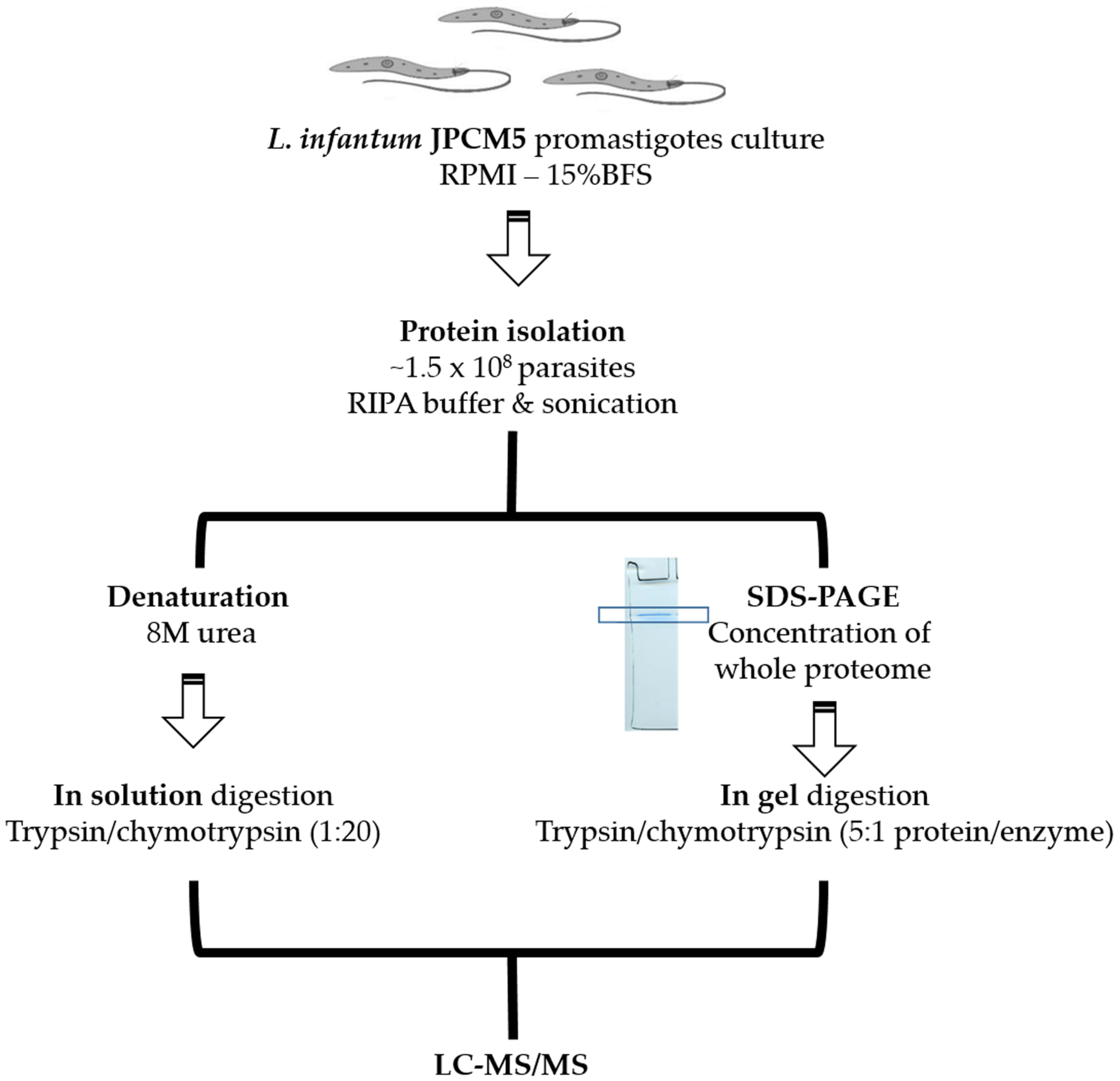
2.1. Leishmania Infantum Culture and Protein Extraction
2.2. In-Gel and In-Solution Digestion of Samples by Trypsin and Chymotrypsin
2.3. Reverse Phase-Liquid Chromatography Mass Spectrometry Analysis (RP-LC-MS/MS)
2.4. Data Analysis
3. Results and Discussion
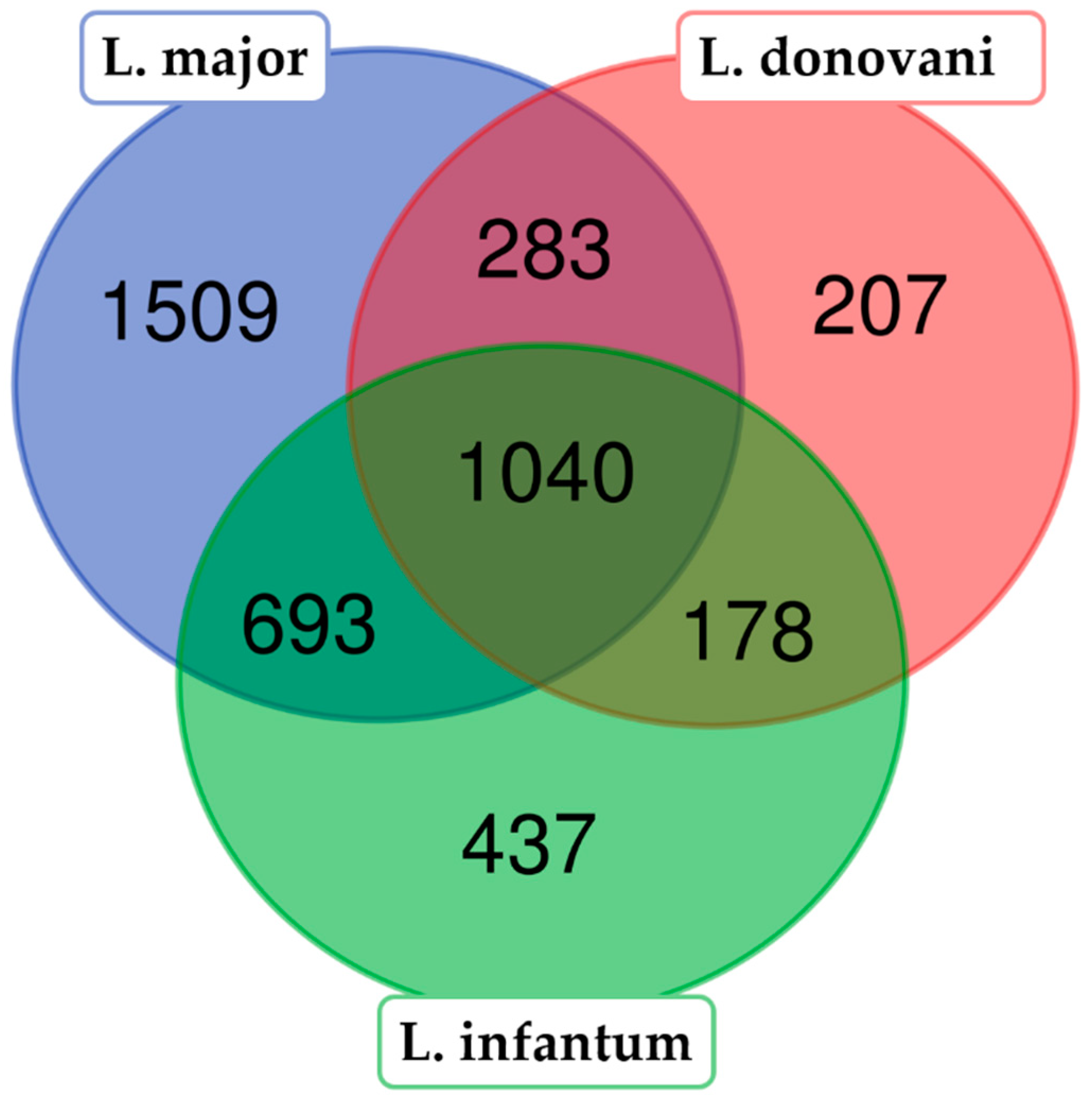
3.1. Protein Identification from the LC−MS/MS Peptide Spectra
3.2. Representativeness of the Translational Machinery and RNA Binding Proteins in the L. infantum Experimental Proteome
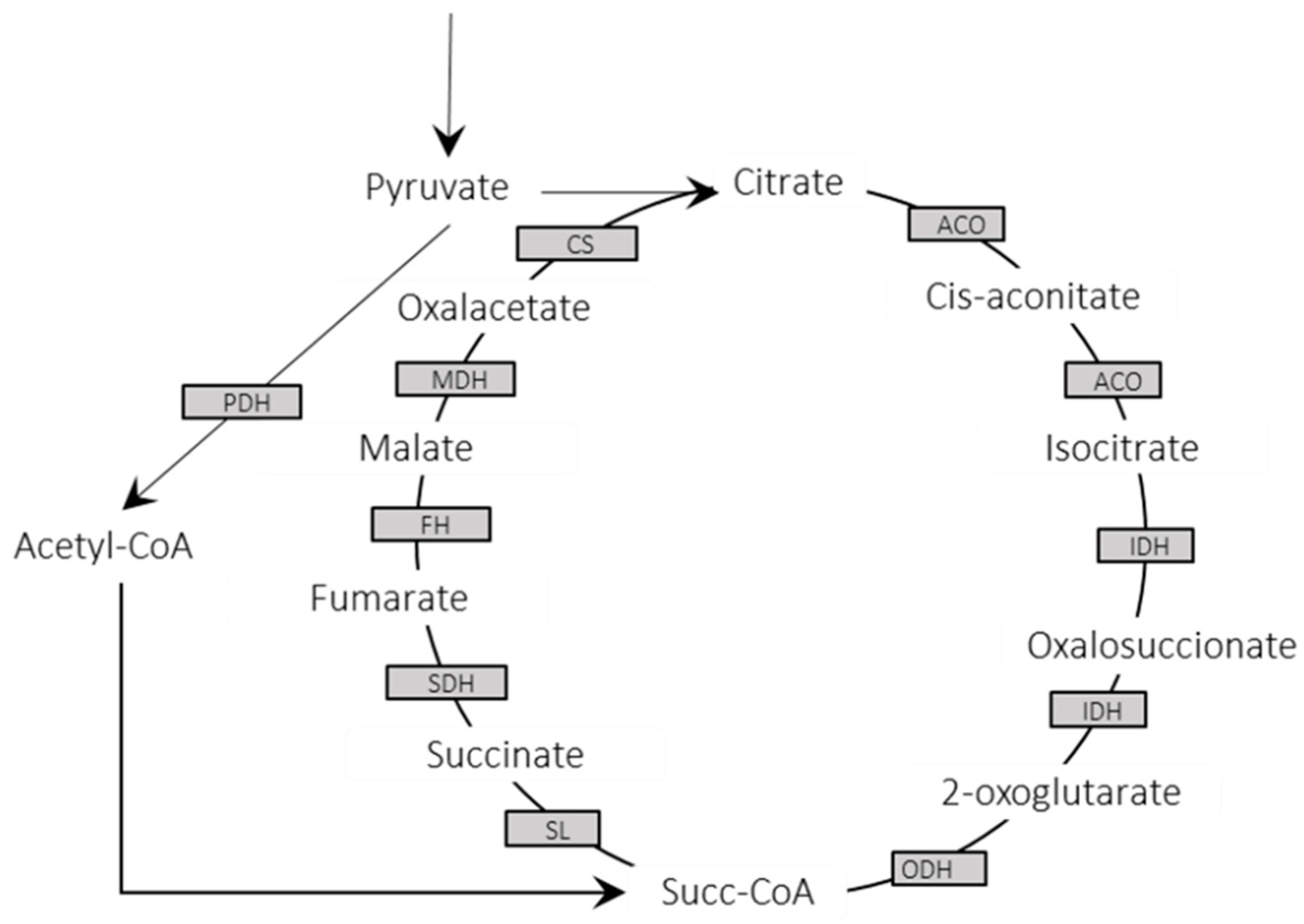
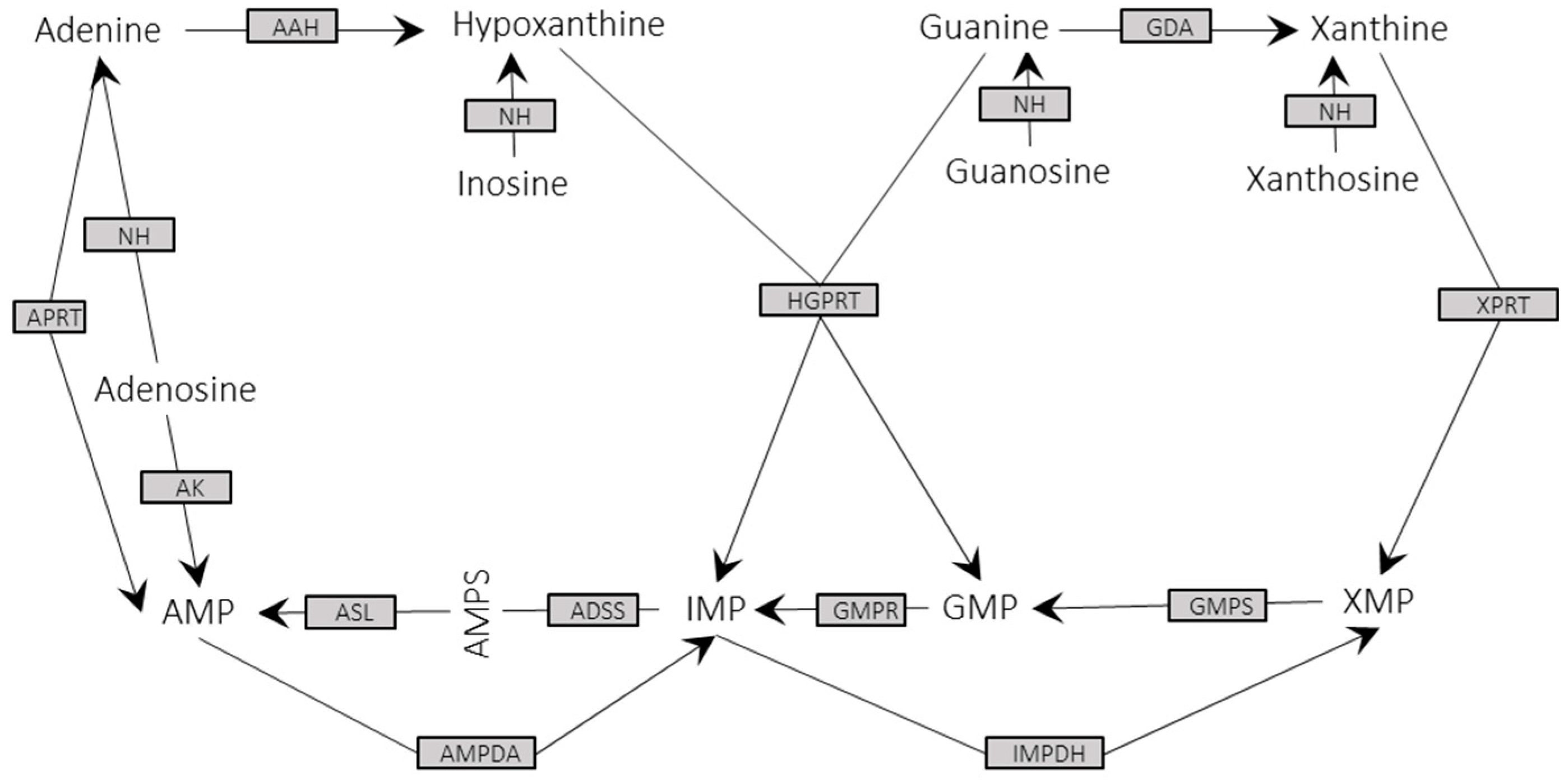
3.3. Metabolic Enzymes and Pathways
3.4. Components of the Proteostasis Network
3.5. Glycosomal Proteins Represent a Substantial Fraction of the Experimentally Detected Proteins in the L. infantum Promastigote
3.6. Exoproteome Components Identified in the L. infantum Experimental Proteome
3.7. Other Relevant Proteins Identified in the L. infantum Promastigote Proteome
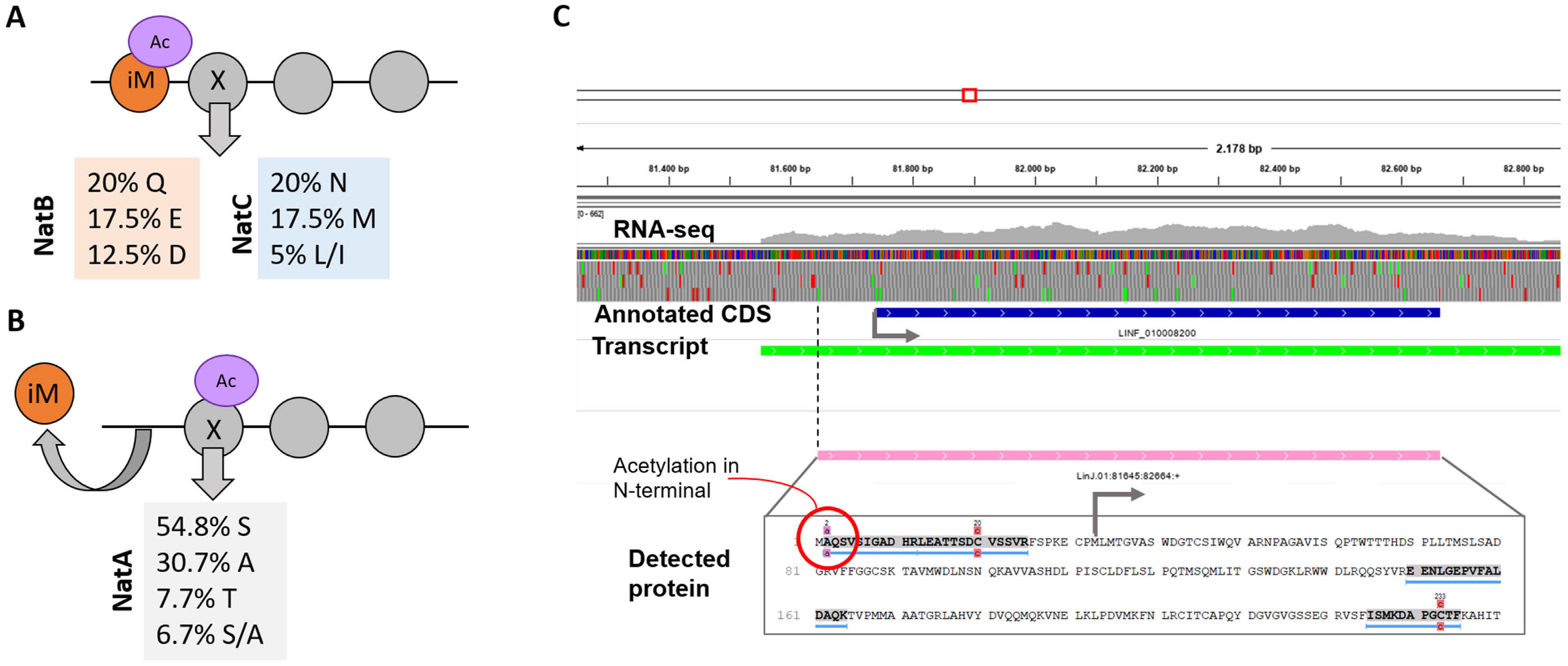
3.8. Detection of Post-Translational Modifications
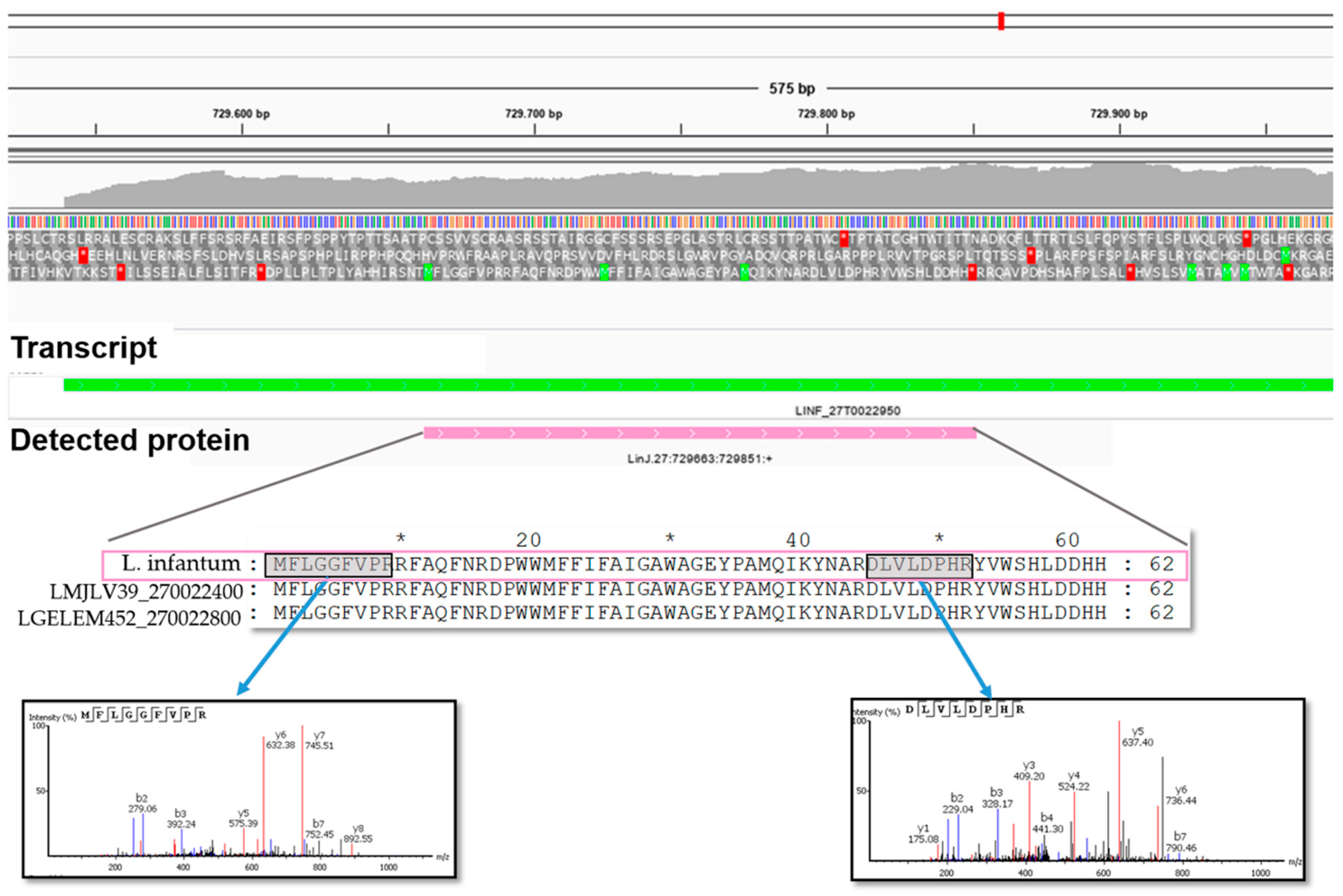
3.9. Proteogenomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Leishmaniasis: Disease, Epidemiology, Diagnosis, Detection and Surveillance, Vector Control, Access to Medicines and Information Resources; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Lukes, J.; Mauricio, I.L.; Schonian, G.; Dujardin, J.-C.; Soteriadou, K.; Dedet, J.-P.; Kuhls, K.; Tintaya, K.W.Q.; Jirku, M.; Chocholova, E.; et al. Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. Proc. Natl. Acad. Sci. USA 2007, 104, 9375–9380. [Google Scholar] [CrossRef] [PubMed]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Berriman, M.; Sisk, E.; Rajandream, M.; Adlem, E.; Anupama, A.; Apostolou, Z.; et al. The genome of the kinetoplastid parasite, Leishmania major. Science 2006, 309, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Downing, T.; Imamura, H.; Decuypere, S.; Clark, T.G.; Coombs, G.H.; Cotton, J.A.; Hilley, J.D.; de Doncker, S.; Maes, I.; Mottram, J.C.; et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011, 21, 2143–2156. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Srivastava, S.; Singh, A.; Singh, S. De novo whole-genome sequence and annotation of a Leishmania strain isolated from a case of post-kala-azar dermal leishmaniasis. Genome Announc. 2015, 3, 4–5. [Google Scholar] [CrossRef]
- Llanes, A.; Restrepo, C.M.; Del Vecchio, G.; Anguizola, F.J.; Lleonart, R. The genome of Leishmania panamensis: Insights into genomics of the L. (Viannia) subgenus. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Raymond, F.; Boisvert, S.; Roy, G.; Ritt, J.F.; Légaré, D.; Isnard, A.; Stanke, M.; Olivier, M.; Tremblay, M.J.; Papadopoulou, B.; et al. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 2012, 40, 1131–1147. [Google Scholar] [CrossRef]
- Real, F.; Vidal, R.O.; Carazzolle, M.F.; Mondego, J.M.C.; Costa, G.G.L.; Herai, R.H.; Würtele, M.; De Carvalho, L.M.E.; Ferreira, R.C.; Mortara, R.A.; et al. The genome sequence of leishmania (Leishmania) amazonensis: Functional annotation and extended analysis of gene models. DNA Res. 2013, 20, 567–581. [Google Scholar] [CrossRef]
- Forsdyke, D.R. Evolutionary Bioinformatics; Springer: New York, NY, USA, 2006; pp. 1–424. [Google Scholar]
- Imamura, H.; Downing, T.; Van den Broeck, F.; Sanders, M.J.; Rijal, S.; Sundar, S.; Mannaert, A.; Vanaerschot, M.; Berg, M.; De Muylder, G.; et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. Elife 2016, 5, e12613. [Google Scholar] [CrossRef]
- Coughlan, S.; Mulhair, P.; Sanders, M.; Schonian, G.; Cotton, J.A.; Downing, T. The genome of Leishmania adleri from a mammalian host highlights chromosome fission in Sauroleishmania. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- González-De La Fuente, S.; Peiró-Pastor, R.; Rastrojo, A.; Moreno, J.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci. Rep. 2017, 7, 18050. [Google Scholar]
- Alonso, G.; Rastrojo, A.; López-Pérez, S.; Requena, J.M.; Aguado, B. Resequencing and assembly of seven complex loci to improve the Leishmania major (Friedlin strain) reference genome. Parasites Vectors 2016, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- González-De La Fuente, S.; Camacho, E.; Peiró-Pastor, R.; Rastrojo, A.; Carrasco-Ramiro, F.; Aguado, B.; Requena, J.M. Complete and de novo assembly of the Leishmania braziliensis (M2904) genome. Mem. Inst. Oswaldo Cruz 2019, 114, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.M.; Alcolea, P.J.; Alonso, A.; Larraga, V. Omics approaches for understanding gene expression in Leishmania: Clues for tackling leishmaniasis. In Protozoan Parasitism: From Omics to Prevention and Control; Pablos-Torró, L.M., Lorenzo-Morales, J., Eds.; Caister Academic Press: Norfolk, UK, 2018; pp. 77–112. [Google Scholar]
- Capelli-Peixoto, J.; Mule, S.N.; Tano, F.T.; Palmisano, G.; Stolf, B.S. Proteomics and Leishmaniasis: Potential clinical applications. Proteom. Clin. Appl. 2019, 13, 1800136. [Google Scholar] [CrossRef]
- Nugent, P.G.; Karsani, S.A.; Wait, R.; Tempero, J.; Smith, D.F. Proteomic analysis of Leishmania mexicana differentiation. Mol. Biochem. Parasitol. 2004, 136, 51–62. [Google Scholar] [CrossRef]
- Leifso, K.; Cohen-Freue, G.; Dogra, N.; Murray, A.; McMaster, W.R. Genomic and proteomic expression analysis of Leishmania promastigote and amastigote life stages: The Leishmania genome is constitutively expressed. Mol. Biochem. Parasitol. 2007, 152, 35–46. [Google Scholar] [CrossRef]
- Rosenzweig, D.; Smith, D.; Opperdoes, F.; Stern, S.; Olafson, R.W.; Zilberstein, D. Retooling Leishmania metabolism: From sand fly gut to human macrophage. FASEB J. 2008, 22, 590–602. [Google Scholar] [CrossRef]
- Jardim, A.; Hardie, D.B.; Boitz, J.; Borchers, C.H. Proteomic profiling of Leishmania donovani promastigote subcellular organelles. J. Proteome Res. 2018, 17, 1194–1215. [Google Scholar] [CrossRef]
- Tasbihi, M.; Shekari, F.; Hajjaran, H.; Masoori, L.; Hadighi, R. Mitochondrial proteome profiling of Leishmania tropica. Microb. Pathog. 2019, 133, 103542. [Google Scholar] [CrossRef]
- Armengaud, J. Proteogenomics and systems biology: Quest for the ultimate missing parts. Expert Rev. Proteom. 2010, 7, 65–77. [Google Scholar] [CrossRef]
- Moreno, M.-L.; Escobar, J.; Izquierdo-Alvarez, A.; Gil, A.; Perez, S.; Pereda, J.; Zapico, I.; Vento, M.; Sabater, L.; Marina, A.; et al. Disulfide stress: A novel type of oxidative stress in acute pancreatitis. Free Radic. Biol. Med. 2014, 70, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.L.; Cantero, A.; del Valle, M.; Marina, A.; Lopez-Gallego, F.; Guisan, J.M.; Berenguer, J.; Hidalgo, A. Engineering the substrate specificity of a thermophilic penicillin acylase from Thermus thermophilus. Appl. Environ. Microbiol. 2013, 79, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteom. 2012, 11, M111.010587. [Google Scholar] [CrossRef] [PubMed]
- Yonghua, H.; Bin, M.; Zaizhong, Z. SPIDER: Software for protein identification from sequence tags with de novo sequencing error. In Proceedings of the IEEE Computational Systems Bioinformatics Conference, Stanford, CA, USA, 19 August 2004. [Google Scholar]
- Han, X.; He, L.; Xin, L.; Shan, B.; Ma, B. PeaksPTM: Mass spectrometry-based identification of peptides with unspecified modifications. J. Proteome Res. 2011, 10, 2930–2936. [Google Scholar] [CrossRef] [PubMed]
- McNicoll, F.; Drummelsmith, J.; Müller, M.; Madore, É.; Boilard, N.; Ouellette, M.; Papadopoulou, B. A combined proteomic and transcriptomic approach to the study of stage differentiation in Leishmania infantum. Proteomics 2006, 6, 3567–3581. [Google Scholar] [CrossRef] [PubMed]
- Foucher, A.L.; Papadopoulou, B.; Ouellette, M. Prefractionation by digitonin extraction increases representation of the cytosolic and intracellular proteome of Leishmania infantum. J. Proteome Res. 2006, 5, 1741–1750. [Google Scholar] [CrossRef]
- Brotherton, M.-C.; Racine, G.; Foucher, A.L.; Drummelsmith, J.; Papadopoulou, B.; Ouellette, M. Analysis of stage-specific expression of basic proteins in Leishmania infantum. J. Proteome Res. 2010, 9, 3842–3853. [Google Scholar] [CrossRef]
- Alcolea, P.J.; Alonso, A.; Larraga, V. Proteome profiling of Leishmania infantum promastigotes. J. Eukaryot. Microbiol. 2011, 58, 352–358. [Google Scholar] [CrossRef]
- Braga, M.S.; Neves, L.X.; Campos, J.M.; Roatt, B.M.; de Oliveira Aguiar Soares, R.D.; Braga, S.L.; de Melo Resende, D.; Reis, A.B.; Castro-Borges, W. Shotgun proteomics to unravel the complexity of the Leishmania infantum exoproteome and the relative abundance of its constituents. Mol. Biochem. Parasitol. 2014, 195, 43–53. [Google Scholar] [CrossRef]
- Santarém, N.; Racine, G.; Silvestre, R.; Cordeiro-da-Silva, A.; Ouellette, M. Exoproteome dynamics in Leishmania infantum. J. Proteom. 2013, 84, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Pawar, H.; Renuse, S.; Khobragade, S.N.; Chavan, S.; Sathe, G.; Kumar, P.; Mahale, K.N.; Gore, K.; Kulkarni, A.; Dixit, T.; et al. Neglected tropical diseases and omics science: Proteogenomics analysis of the promastigote stage of leishmania major parasite. Omi. A J. Integr. Biol. 2014, 18, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Nirujogi, R.S.; Pawar, H.; Renuse, S.; Kumar, P.; Chavan, S.; Sathe, G.; Sharma, J.; Khobragade, S.; Pande, J.; Modak, B.; et al. Moving from unsequenced to sequenced genome: Reanalysis of the proteome of Leishmania donovani. J. Proteom. 2014, 97, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Pawar, H.; Sahasrabuddhe, N.A.; Renuse, S.; Keerthikumar, S.; Sharma, J.; Kumar, G.S.S.; Venugopal, A.; Sekhar, N.R.; Kelkar, D.S.; Nemade, H.; et al. A proteogenomic approach to map the proteome of an unsequenced pathogen—Leishmania donovani. Proteomics 2012, 12, 832–844. [Google Scholar] [CrossRef]
- Ramrath, D.J.F.; Niemann, M.; Leibundgut, M.; Bieri, P.; Prange, C.; Horn, E.K.; Leitner, A.; Boehringer, D.; Schneider, A.; Ban, N. Evolutionary shift toward protein-based architecture in trypanosomal mitochondrial ribosomes. Science 2018, 362, eaau7735. [Google Scholar] [CrossRef]
- Clayton, C.; Shapira, M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 2007, 156, 93–101. [Google Scholar] [CrossRef]
- Requena, J.M. Lights and shadows on gene organization and regulation of gene expression in Leishmania. Front. Biosci. Landmark Ed. 2011, 16, 2069–2085. [Google Scholar] [CrossRef]
- Beckmann, B.M.; Castello, A.; Medenbach, J. The expanding universe of ribonucleoproteins: Of novel RNA-binding proteins and unconventional interactions. Pflugers Arch. Eur. J. Physiol. 2016, 468, 1029–1040. [Google Scholar] [CrossRef]
- Kramer, S.; Carrington, M. Trans-acting proteins regulating mRNA maturation, stability and translation in trypanosomatids. Trends Parasitol. 2011, 27, 23–30. [Google Scholar] [CrossRef][Green Version]
- De Gaudenzi, J.G.; Frasch, A.C.; Clayton, C. RNA-binding domain proteins in Kinetoplastids: A comparative analysis. Eukaryot. Cell 2014, 4, 2106–2114. [Google Scholar] [CrossRef]
- Nandan, D.; Thomas, S.A.; Nguyen, A.; Moon, K.M.; Foster, L.J.; Reiner, N.E. Comprehensive identification of mRNABinding proteins of Leishmania donovani by interactome capture. PLoS ONE 2017, 12, e0170068. [Google Scholar] [CrossRef] [PubMed]
- Folgueira, C.; Martínez-Bonet, M.; Requena, J.M. The Leishmania infantum PUF proteins are targets of the humoral response during visceral leishmaniasis. BMC Res. Notes 2010, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Jhawar, J.; Sarkar, R.R. Dissecting Leishmania infantum energy metabolism—A systems perspective. PLoS ONE 2015, 10, e0137976. [Google Scholar] [CrossRef] [PubMed]
- Colasante, C.; Voncken, F.; Manful, T.; Ruppert, T.; Tielens, A.G.M.; van Hellemond, J.J.; Clayton, C. Proteins and lipids of glycosomal membranes from Leishmania tarentolae and Trypanosoma brucei. F1000Research 2013, 2, 1–15. [Google Scholar] [CrossRef]
- Camacho, E.; Rastrojo, A.; Sanchiz, Á.; González-de la Fuente, S.; Aguado, B.; Requena, J.M. Leishmania mitochondrial genomes: Maxicircle structure and heterogeneity of minicircles. Genes (Basel) 2019, 10, 758. [Google Scholar] [CrossRef]
- Paugam, A.; Bulteau, A.L.; Dupouy-Camet, J.; Creuzet, C.; Friguet, B. Characterization and role of protozoan parasite proteasomes. Trends Parasitol. 2003, 19, 55–59. [Google Scholar] [CrossRef]
- Silva-Jardim, I.; Fátima Horta, M.; Ramalho-Pinto, F.J. The Leishmania chagasi proteasome: Role in promastigotes growth and amastigotes survival within murine macrophages. Acta Trop. 2004, 91, 121–130. [Google Scholar] [CrossRef]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.N.; Myburgh, E.; Gao, M.-Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Requena, J.M.; Montalvo, A.M.; Fraga, J. Molecular chaperones of Leishmania: Central players in many stress-related and -unrelated physiological processes. Biomed Res. Int. 2015, 2015, 1–21. [Google Scholar] [CrossRef]
- Bauer, S.; Morris, M.T. Glycosome biogenesis in trypanosomes and the de novo dilemma. PLoS Negl. Trop. Dis. 2017, 11, e0005333. [Google Scholar] [CrossRef] [PubMed]
- Pilar, A.V.C.; Strasser, R.; McLean, J.; Quinn, E.; Cyr, N.; Hojjat, H.; Kottarampatel, A.H.; Jardim, A. Analysis of the Leishmania peroxin 7 interactions with peroxin 5, peroxin 14 and PTS2 ligands. Biochem. J. 2014, 460, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Jamdhade, M.D.; Pawar, H.; Chavan, S.; Sathe, G.; Umasankar, P.K.; Mahale, K.N.; Dixit, T.; Madugundu, A.K.; Prasad, T.S.K.; Gowda, H.; et al. Comprehensive Proteomics analysis of glycosomes from Leishmania donovani. OMICS A J. Integr. Biol. 2015, 19, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Boitz, J.M.; Ullman, B.; Jardim, A.; Carter, N.S. Purine salvage in Leishmania: Complex or simple by design? Trends Parasitol. 2012, 28, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, K.; Dubey, V.K. Fresh insights into the pyrimidine metabolism in the trypanosomatids. Parasites Vectors 2018, 11, 1–15. [Google Scholar] [CrossRef]
- Pérez-Cabezas, B.; Santarém, N.; Cecílio, P.; Silva, C.; Silvestre, R.; AM Catita, J.; Cordeiro da Silva, A. More than just exosomes: Distinct Leishmania infantum extracellular products potentiate the establishment of infection. J. Extracell. Vesicles 2019, 8, 1541708. [Google Scholar] [CrossRef]
- Atayde, V.D.; Aslan, H.; Townsend, S.; Hassani, K.; Kamhawi, S.; Olivier, M. Exosome secretion by the parasitic protozoan Leishmania within the sand fly midgut. Cell Rep. 2015, 13, 957–967. [Google Scholar] [CrossRef]
- Avilán, L.; Gualdrón-López, M.; Quiñones, W.; González-González, L.; Hannaert, V.; Michels, P.A.M.; Concepción, J.-L. Enolase: A key player in the metabolism and a probable virulence factor of Trypanosomatid parasites—Perspectives for its use as a therapeutic target. Enzyme Res. 2011, 2011, 932549. [Google Scholar] [CrossRef]
- Brotherton, M.C.; Racine, G.; Ouameur, A.A.; Leprohon, P.; Papadopoulou, B.; Ouellette, M. Analysis of membrane-enriched and high molecular weight proteins in Leishmania infantum promastigotes and axenic amastigotes. J. Proteome Res. 2012, 11, 3974–3985. [Google Scholar] [CrossRef]
- Beneke, T.; Demay, F.; Hookway, E.; Ashman, N.; Jeffery, H.; Smith, J.; Valli, J.; Becvar, T.; Myskova, J.; Lestinova, T.; et al. Genetic dissection of a Leishmania flagellar proteome demonstrates requirement for directional motility in sand fly infections. PLoS Pathog. 2019, 15, e1007828. [Google Scholar] [CrossRef]
- Morales, M.A.; Watanabe, R.; Dacher, M.; Chafey, P.; Osorio, Y.; Fortéa, J.; Scott, D.A.; Beverley, S.M.; Ommen, G.; Clos, J.; et al. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc. Natl. Acad. Sci. USA 2010, 107, 8381–8386. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, D.; Smith, D.; Myler, P.J.; Olafson, R.W.; Zilberstein, D. Post-translational modification of cellular proteins during Leishmania donovani differentiation. Proteomics 2008, 8, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.S.; Branquinha, M.H.; D’Avila-Levy, C.M.; Kneipp, L.; Sodré, C.L. Proteins and Proteomics of Leishmania and Trypanosoma; Springer: New York, NY, USA, 2014; ISBN 978-94-007-7304-2. [Google Scholar]
- Tsigankov, P.; Gherardini, P.F.; Helmer-Citterich, M.; Späth, G.F.; Zilberstein, D. Phosphoproteomic analysis of differentiating Leishmania parasites reveals a unique stage-specific phosphorylation motif. J. Proteome Res. 2013, 12, 3405–3412. [Google Scholar] [CrossRef] [PubMed]
- Sprung, R.; Chen, Y.; Zhang, K.; Cheng, D.; Zhang, T.; Peng, J.; Zhao, Y. Identification and validation of eukaryotic aspartate and glutamate methylation in proteins. J. Proteome Res. 2008, 7, 1001–1006. [Google Scholar] [CrossRef]
- Alonso, V.L.; Serra, E.C. Lysine acetylation: Elucidating the components of an emerging global signaling pathway in trypanosomes. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Thomas, S.; Green, A.; Sturm, N.R.; Campbell, D.A.; Myler, P.J. Histone acetylations mark origins of polycistronic transcription in Leishmania major. BMC Genom. 2009, 10, 152. [Google Scholar] [CrossRef]
- Respuela, P.; Ferella, M.; Rada-Iglesias, A.; Aslund, L. Histone acetylation and methylation at sites initiating divergent polycistronic transcription in Trypanosoma cruzi. J. Biol. Chem. 2008, 283, 15884–15892. [Google Scholar] [CrossRef]
- Cuervo, P.; Domont, G.B.; De Jesus, J.B. Proteomics of trypanosomatids of human medical importance. J. Proteom. 2010, 73, 845–867. [Google Scholar] [CrossRef]
- Linster, E.; Wirtz, M. N-terminal acetylation: An essential protein modification emerges as an important regulator of stress responses. J. Exp. Bot. 2018, 69, 4555–4568. [Google Scholar] [CrossRef]
- Gupta, N.; Tanner, S.; Jaitly, N.; Adkins, J.N.; Lipton, M.; Edwards, R.; Romine, M.; Osterman, A.; Bafna, V.; Smith, R.D.; et al. Whole proteome analysis of post-translational modifications: Applications of mass-spectrometry for proteogenomic annotation. Genome Res. 2007, 17, 1362–1377. [Google Scholar] [CrossRef][Green Version]
- Manzano-Román, R.; Fuentes, M. Relevance and proteomics challenge of functional posttranslational modifications in Kinetoplastid parasites. J. Proteom. 2020, 220, 103762. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Fahlman, R.P.; Esmaili, M.; Fon, E.A. Formylation of eukaryotic cytoplasmic proteins: Linking stress to degradation. Trends Biochem. Sci. 2019, 44, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Nε-Formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2008, 36, 570–577. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Unique Peptides | Description |
|---|---|---|
| LINF_040016700 | 12 | Fructose-1-6-bisphosphatase |
| LINF_120010600 | 43 | Glucose-6-phosphate isomerase |
| LINF_200006000 | 41 | Phosphoglycerate kinase C-glycosomal |
| LINF_210007800 | 47 | Hexokinase |
| LINF_210012000 | 13 | Phosphoglucomutase |
| LINF_230009500 | 10 | Aldose 1-epimerase-like protein |
| LINF_240013700 | 32 | Triosephosphate isomerase |
| LINF_250017300 | 46 | Aldehyde dehydrogenase—mitochondrial precursor |
| LINF_270024900 | 56 | Glycosomal phosphoenolpyruvate carboxykinase |
| LINF_290032900 | 23 | ATP-dependent phosphofructokinase |
| LINF_300035000 | 49 | Glyceraldehyde 3-phosphate dehydrogenase—glycosomal |
| LINF_300039500 | 13 | PAS-domain containing phosphoglycerate kinase |
| LINF_340040800 | 13 | Aldose 1-epimerase-like protein |
| Gene ID | Unique Peptides | Description |
|---|---|---|
| LINF_140018000 | 55 | Enolase |
| LINF_180019200 | 23 | Pyruvate dehydrogenase e1 component α subunit |
| LINF_210011100 | 10 | Dihydrolipoamide acetyltransferase |
| LINF_210012000 | 13 | Phosphoglucomutase |
| LINF_230009000 | 43 | NADP-dependent alcohol dehydrogenase |
| LINF_230014800 | 47 | Acetyl-CoA synthetase |
| LINF_250023800 | 29 | Pyruvate dehydrogenase e1 β subunit |
| LINF_290025700 | 4 | Dihydrolipoamide dehydrogenase |
| LINF_310034500 | 2 | Dihydrolipoamide dehydrogenase |
| LINF_320040600 | 37 | Dihydrolipoamide dehydrogenase |
| LINF_350005300 LINF_350005400 | 43 | Pyruvate kinase |
| LINF_360030600 | 43 | Glyceraldehyde 3-phosphate dehydrogenase—cytosolic |
| LINF_360034400 | 19 | Dihydrolipoamide acetyltransferase precursor |
| Gene ID | Unique Peptides | Molecular Chaperone |
|---|---|---|
| LINF_260011400 | 9 | Heat shock protein 10 (HSP10) |
| LINF_320040500 | 5 | HSP40/JDP45 |
| LINF_350035100 | 7 | HSP40/JDP50 |
| LINF_360027100 | 19 | HSP60/cpn60.2 |
| LINF_360027200 | 26 | HSP60/cpn60.3 |
| LINF_240010000 | 5 | HSP40/JDP8 |
| LINF_260017400 | 42 | HSP70.4 |
| LINF_280017800 | 51 | Grp78/BiP |
| LINF_330033000 | 48 | HSP75/TRAP-1 |
| LINF_020012400 | 23 | HSP78 |
| LINF_330009000 | 22 | HSP83/90 |
| Gene ID | Unique Peptides | Description |
|---|---|---|
| LINF_060011200 | 7 | Deoxyuridine triphosphatase |
| LINF_160010400 | 5 | Dihydroorotate dehydrogenase (fumarate) |
| LINF_160010500 | 15 | Aspartate carbamoyltransferase |
| LINF_160010700 | 28 | Orotate phosphoribosyltransferase |
| LINF_160011200 | 6 | Carbamoyl-phosphate synthase |
| LINF_180021400 | 17 | Nonspecific nucleoside hydrolase |
| LINF_340016700 | 8 | Uracil phosphoribosyltransferase |
| Gene ID | Description | Features [Ref.] |
|---|---|---|
| LINF 050017500; LINF 040007000 | Surface antigens | Virulence factor [61] |
| LINF_090013900 | Oligopeptidase b | Virulence factor [35] |
| LINF_100010100 | GP63-leishmanolysin | Virulence factor [60,61] |
| LINF_120014700 | Surface antigen protein 2 | Virulence factor [61] |
| LINF_140018000 | Enolase | Virulence factor [35,62] |
| LINF_150019000 | Tryparedoxin peroxidase | Virulence factor [61] |
| LINF_170005900 | Elongation factor 1-α | Exosome marker [61] |
| LINF_190020600 | Cysteine peptidase A (CPA) | Virulence factor [61] |
| LINF_200018000 | Calpain-like cysteine peptidase | Virulence factor [61] |
| LINF_280035000 LINF_280036000 | HSP70 | Exosome marker [61] |
| LINF_280034700 | Receptor for activated C kinase 1 | Immunomodulator [35] |
| LINF_320036700 | Nucleoside diphosphate kinase b | Immunomodulator [35] |
| LINF_330009000 | HSP83/90 | Exosome marker [61] |
| LINF_350027300 LINF_350027500 | Kinetoplastid membrane protein 11 (KMP11) | Immunomodulator [35] |
| LINF_360018400 | Fructose-1-6-bisphosphate aldolase | Immunomodulator [60] |
| Gene ID | Description | Position |
|---|---|---|
| LINF_040005600 | Hypothetical protein-conserved | T187 |
| LINF_130007700; LINF_130007800; LINF_130008000; LINF_130008200; LINF_130008300; LINF_130008400; LINF_130008600; LINF_130008700 | α tubulin | T334; Y357 |
| LINF_180007700 | Glycogen synthase kinase 3 (GSK-3) | Y186 |
| LINF_190017000 | Hypothetical protein-conserved | S120 |
| LINF_200011800 | rRNA biogenesis protein-like protein | Y571 |
| LINF_220013200 | Hypothetical protein-conserved | S45 |
| LINF_230014600 | 3-ketoacyl-CoA thiolase | S229 |
| LINF_280035400 | HSP70 | T159; T164 |
| LINF_360015600; LINF_360015700 | 40S ribosomal protein S10 | S157 |
| LINF_360068400 | Flagellum targeting protein KHARON1 | S158 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchiz, Á.; Morato, E.; Rastrojo, A.; Camacho, E.; González-de la Fuente, S.; Marina, A.; Aguado, B.; Requena, J.M. The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations. Genes 2020, 11, 1036. https://doi.org/10.3390/genes11091036
Sanchiz Á, Morato E, Rastrojo A, Camacho E, González-de la Fuente S, Marina A, Aguado B, Requena JM. The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations. Genes. 2020; 11(9):1036. https://doi.org/10.3390/genes11091036
Chicago/Turabian StyleSanchiz, África, Esperanza Morato, Alberto Rastrojo, Esther Camacho, Sandra González-de la Fuente, Anabel Marina, Begoña Aguado, and Jose M. Requena. 2020. "The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations" Genes 11, no. 9: 1036. https://doi.org/10.3390/genes11091036
APA StyleSanchiz, Á., Morato, E., Rastrojo, A., Camacho, E., González-de la Fuente, S., Marina, A., Aguado, B., & Requena, J. M. (2020). The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations. Genes, 11(9), 1036. https://doi.org/10.3390/genes11091036