Abstract
Embryonal tumor with multilayered rosettes (ETMR) is an aggressive and rare pediatric embryonal brain tumor. Amplification of C19MC microRNA cluster and expression of LIN28 are distinctive features of ETMR. Despite the increasing efforts to decipher ETMR, the biology remains poorly understood. To date, the role of aberrant alternative splicing in ETMR has not been thoroughly investigated. In the current study, a comprehensive analysis was performed on published unprocessed RNA-seq reads of tissue-matched ETMR and fetal controls datasets. Gene expression was quantified in samples using Kallisto/sleuth pipeline. For the alternative splicing analysis, STAR, SplAdder and rMATS were used. Functional enrichment analysis was subsequently performed using Metascape. The expression analysis identified a total of 3622 differentially expressed genes (DEGs) between ETMR and fetal controls while 1627 genes showed differential alternative splicing patterns. Interestingly, genes with significant alternative splicing events in ETMR were identified to be involved in signaling pathways such as ErbB, mTOR and MAPK pathways as well as ubiquitin-mediated proteolysis, cell cycle and autophagy. Moreover, up-regulated DEGs with alternative splicing events were involved in important biological processes including nuclear transport, regulation of cell cycle and regulation of Wnt signaling pathway. These findings highlight the role of aberrant alternative splicing in shaping the ETMR tumor landscape, and the identified pathways constitute potential therapeutic targets.
1. Introduction
Embryonal tumor with multilayered rosettes (ETMR) is a rare and aggressive pediatric embryonal brain tumor affecting infants and young children less than 4 years of age [1,2]. Despite intensive multimodal treatment, ETMR is a disease of poor prognosis with only 20% long-term survival [3,4,5,6,7,8,9]. ETMRs are mostly characterized by two distinctive molecular markers; amplification of Chr19q13.41 miRNA cluster (C19MC) [10] and high expression of LIN28A; a pluripotency factor and RNA-binding protein. Furthermore, the overexpression of a specific isoform of DNA methyltransferase 3 β (DNMT3B) was observed in ETMR [11]. High expression of LIN28A is implicated in neural development and pathogenesis of other advanced cancers [12]. Furthermore, the role of LIN28A in regulating splicing and gene expression programs has been investigated in cancer. For example, LIN28A was reported to cause significant isoform switches in genes involved in breast cancer biology [13]. In addition, LIN28A was demonstrated to bind to messenger RNAs (mRNAs) at specific motifs and regulates protein abundance of splicing regulators [14]. Despite the increasing efforts to decipher ETMR, an adequate understanding of the biology remains elusive. It is well established that alternative splicing (AS) is a key mechanism of post-transcriptional regulation of protein-coding genes that enables a single gene to produce multiple proteins [15].
Growing evidence has revealed that aberrant AS is one of the important hallmarks in cancer [16]. Through cellular plasticity that is offered by AS, cancer cells can produce certain protein isoforms favoring tumor growth and allow adaptation to their microenvironment [17,18]. Abnormal AS was also demonstrated to affect nearly all aspects of tumor biology, including cell cycle control, invasion, metastasis, angiogenesis, metabolism apoptosis, and drug resistance [19,20,21]. Many aberrant splicing events and variations in the abundance of alternatively spliced transcripts have been reported in different cancers including breast, pancreatic, liver, and multiple myeloma [22,23,24,25,26]. Thus, the potential role of AS in defining a new therapeutic target is being increasingly investigated. Nevertheless, the role of aberrant AS in ETMR remains largely unexplored.
In the current study, a comprehensive analysis was performed on published RNA-sequencing reads of ETMR and tissue-matched fetal controls from the Human Developmental Biology Resource (HDBR) datasets to identify and investigate the role of alternative splicing in ETMR. Our study showed that top up-regulated differentially expressed genes were involved in RNA splicing and processing, ubiquitination, and autophagy. Moreover, genes with AS events in ETMR were involved in developmental processes as well as cancer-related signaling pathways including ErbB, mTOR, and MAPK pathways. Finally, our work demonstrates the potential role of aberrant AS in the biology of ETMR.
2. Materials and Methods
2.1. Publicly Available RNA-Seq Data
Published unprocessed RNA-seq reads of nine ETMR samples were downloaded from NCBI’s Sequence Read Archive (SRA) (dataset accession number: SRP032476) [11], while the unprocessed RNA-seq reads of nine tissue-matched fetal controls from the Human Developmental Biology Resource (HDBR) were downloaded from ArrayExpress Archive (dataset accession number E-MTAB-4840) [27]. Both datasets were sequenced on an Illumina HiSeq 2000 (100-bp paired-end).
2.2. RNA-Seq Data Analysis
Quality control processes including adapter trimming, low-quality bases and short reads removal were performed on all the 18 fastq files using fastp software [28]. Links to QC reports are provided in Table S1. Gene expression was quantified using the Kallisto/sleuth pipeline. Read quantification was performed with Kallisto (v0.46.1), a pseudoalignment-based method to quantify RNA abundance at transcript-level in transcripts per million (TPM) counts [29]. Kallisto quant was utilized with the number of bootstraps set to 100 using ENSEMBL cDNA transcripts (Human assembly hg38 (GRCh38), release 94) for indexing. hierarchical clustering and principal component analysis (PCA) of the samples were performed and the plots are provided in Supplementary Materials (Figures S1 and S2). Downstream differential gene expression was performed using Sleuth R package (v0.30.0) [30] to leverage the bootstrap estimates of Kallisto and to output model-based, gene-level normalized TPM matrix. The normalized values were also corrected for potential batch effects due to RNA-seq data derived from two different sequencing core facilities. For each gene, both the likelihood ratio test (LRT) and Wald test (WT) were performed on the condition parameter to obtain their respective FDR-corrected p-values. Significant genes were those passing the two tests at a cutoff of false discovery rate (FDR) < 0.05. Plot_transcript_heatmap function in Sleuth package was utilized to visualize the cluster analysis. EnhancedVolcano R package was used to generate the volcano plot [31], which is a visual tool for displaying differentially expressed genes (DEGs) among overall gene expression levels. The Venn diagram was generated using InteractiVenn [32].
For the alternative splicing analysis, reads were mapped to the Human assembly hg38 using STAR. Two samples were excluded from the AS analysis (ETMR_9, as it was clustered with the controls and Control_3, as it had a low read count for the subsequent AS analysis). Identification and quantification of alternative splicing events (skipped exon, alternative 5′ splice site, and 3′ splice site, mutually exclusive exons, and retained intron) were carried out using both SplAdder [33] and rMATS (replicate multivariate analysis of transcript splicing) [34]. Only events that were detected by both AS tools in at least half of the ETMR samples compared to the controls at p-value < 0.05 were considered significant. IGV (Integrated Genomic Viewer) was utilized to visualize examples of genes with significant RI events in ETMR [35].
QC and alignment statistics are provided in Table S1.
2.3. Functional Enrichment and PPI Network Analysis
Functional enrichment analysis of differentially expressed and differentially spliced genes was subsequently performed using Metascape. All statistical values reported were corrected for multiple hypothesis testing by Benjamini–Hochberg FDR (q-values), and significant terms are chosen based on FDR < 0.05. STRING [36], and an online biological database and resource for known and predicted protein–protein interactions (PPIs) were used to construct a network of PPI of the DEG-encoded proteins with interaction score > 0.4 (medium confidence). Cytoscape software was then used for visualization of the PPIs network using “yFiles organic layout [37].
3. Results
3.1. Identification of Differentially Expressed Genes (DEGs) between ETMR and Fetal Normal Control
After performing quality control and data normalization, a total of 5027 transcripts (3622 protein-coding genes) were identified by Sleuth. 2599 (2052 genes) were up-regulated and 2428 (1901 genes) were down-regulated, based on the cut-off criteria (adjusted p-value < 0.05 and log2 fold change (FC) between 2.4 and −2.4). Hierarchical cluster analysis demonstrated that the differentially expressed transcripts accurately distinguished ETMR samples from fetal control samples (Figure 1A). Volcano plot was generated to representDEGs with log2 FC scores and −log10 adjusted p-values (Figure 1B).
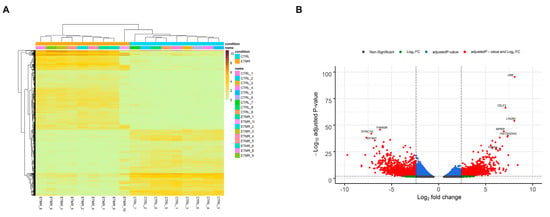
Figure 1.
Overview of the differentially expressed genes (DEGs). (A) Heatmap of the differentially expressed transcripts. Each row read represents a single transcript, and each column represents a sample in each condition. (B) Volcano plot overviews the DEGs and highlighting the top up-regulated and down-regulated genes. The log2 (FC—fold change) is plotted on the x-axis, and the negative log10 adjusted p-value is plotted on the y-axis. The horizontal line represents the cutoff of the adjusted p-value (0.001) and the vertical lines represent the cutoff of the log2 FC (2.4 and −2.4).
3.2. Enrichment Analysis of the DEGs
DEGs were mainly enriched in developmental processes including regulation of cell morphogenesis, regulation of neuron projection development, in addition to histone modification, regulation of autophagy, regulation of mRNA metabolic process, regulation of ubiquitin-dependent protein catabolic process and protein folding (Figure 2) (Table S1). According to the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis, the DEGs were mainly enriched in Alcoholism, Axon guidance, Regulation of actin cytoskeleton, Cell cycle, Hedgehog signaling pathway, MAPK signaling pathway Proteoglycans in cancer and mTOR signaling pathway (Table S1). Most of the enriched biological processes and pathways were obtained from up-regulated genes, while the down-regulated genes made a lesser contribution.
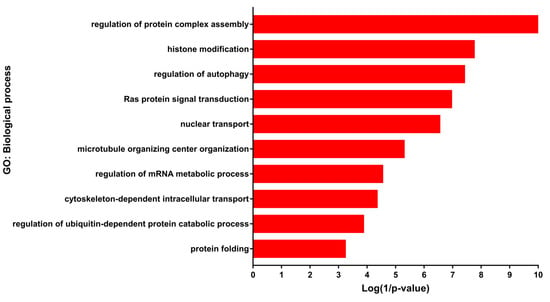
Figure 2.
Gene ontology (GO) enrichment of differentially expressed genes (DEGs). Selected biological processes and their corresponding DEGs.
3.3. PPI Network Analysis of Up-Regulated DEGs
Network analysis of the up-regulated genes produced a PPI network –that is composed of 25 nodes (Table 1) and 68 edges (Figure 3A). PPI network was enriched in RNA splicing (FDR = 3.987 × 10−06), regulation of alternative mRNA splicing, via spliceosome (FDR = 0.00079), protein polyubiquitination (FDR = 0.0079), autophagy of mitochondrion (FDR = 0.025), autophagy (FDR = 0.039), and mitophagy (FDR = 0.015) (Figure 3B)(Table S2).

Table 1.
List of interacting up-regulated protein-coding genes.
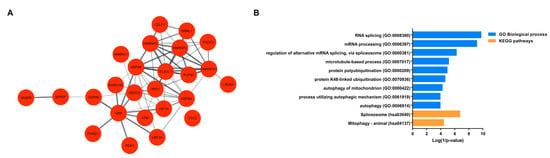
Figure 3.
Enrichment and network analyses of up-regulated DEGs. (A) Protein–protein interaction (PPI) network of selected most up-regulated genes (nodes). The interactions (edges) line thickness indicates the strength of data support. (B) Enrichment analysis of the interacting up-regulated genes in the PPI network. Biological processes (blue) and KEGG pathways (orange) are plotted on the y-axis versus their Log (1/p-values) on the x-axis.
3.4. Alternative Splicing Events in ETMR
A total of 1627 genes were detected to have significant AS events using both Spladder and rMATS tools in ETMR. The distribution of events was as follows: 1023 (63%) genes had SE, 349 (22%) genes had MXE, 115 (7%) genes had A3SS, 86 (5%) genes had A5SS, and 54 (3%) genes showed retained introns (Figure 4A). Interestingly, AS analysis also identified that one gene may undergo more than one type of AS event (Table S3). The significant alternatively spliced genes were enriched in microtubule-based process (FDR = 1 × 10−16), ubiquitin-mediated proteolysis (FDR = 6.3 × 10−07), RNA transport (FDR = 0.005), mRNA processing (FDR = 1 × 10−11), autophagy (FDR = 0.0007), as well as signaling pathways including ErbB signaling pathway (FDR = 0.00019), mTOR signaling pathway (FDR = 0.0025), MAPK signaling pathway (FDR = 0.031) (Figure 3B and Table S3).

Figure 4.
Overview of alternative splicing (AS) in embryonal tumor with multilayered rosettes (ETMR) and enrichment analysis of genes with AS events. (A). Pie chart showing genes’ count with different AS events. (B). Biological processes and KEGG pathways enriched by significant alternatively spliced genes.
3.5. Up-Regulated DEGs with AS Events
A total of 453 up-regulated DEGs were found to have significant AS events (Figure 5A) (Table S4). Enrichment analysis revealed that these genes were enriched in various biological processes and pathways like endocytosis (FDR = 5 × 10−05), histone modification (FDR = 1.2 × 10−06), nuclear transport (FDR = 3.1 × 10−06), ubiquitin-dependent protein catabolic process (FDR = 7.9 × 10−06), regulation of cell cycle process (FDR = 2.5 × 10−05), and regulation of Wnt signaling pathway (FDR = 0.03) (Figure 5B) (Table S4). Two examples of up-regulated genes that displayed intron retention in ETMR are Tetratricopeptide Repeat Domain 3 (TTC3) also known as E3 ubiquitin-protein ligase and exportin-1 (XPO1) (also referred to as CRM1) (Figure 5C). TTC3 showed retention of intron 2, while XPO1 showed retention of intron 12, in 8 out of the 9 ETMR cases.
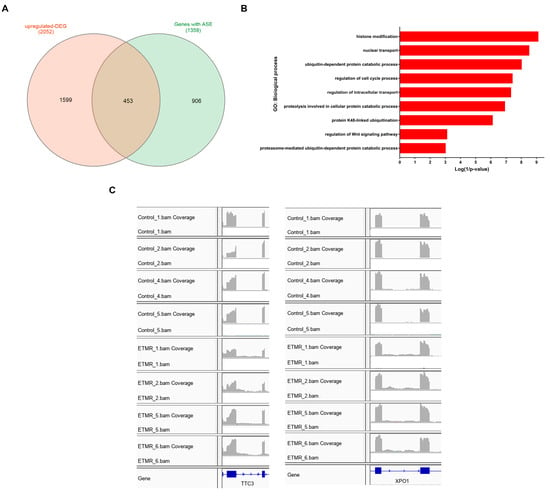
Figure 5.
Enrichment analysis and examples of up-regulated differentially expressed genes (DEGs) with significant AS events. (A). Venn diagram showing common genes between DEGs and genes with significant AS events. (B). Biological processes are enriched by the up-regulated DEGs with significant AS events. (C). IGV(Integrated Genomic Viewer) plots demonstrate RIs in E3 ubiquitin-protein ligase (TTC3) and exportin-1 (XPO1) in ETMR compared to fetal control samples.
4. Discussion
In the current study, we presented an overall transcriptomic analysis of ETMR to gain a better understanding of theirbiology and propose new therapeutic perspectives.Almost all human multi-exon genes are being regulated by AS. Cancer cells are capable of dynamically changing gene expression and favoring the expression of aberrant oncogenic splice variants to overcome stresses within the tumor microenvironment [38]. Abnormal AS is now regarded as a valuable indicator of carcinogenic processes and prognosis, as well as a potential target of treatment in several types of cancer [39,40,41,42,43,44].Not only is it a valuable diagnostic marker of ETMRs [45], LIN28A overexpression can be functionally significant as well. LIN28A was reported as a regulator of self-renewal capacity in cancer stem cells, cellular metabolism, and the cell cycle through binding and repression of let-7 microRNAs [12,46,47]. Our enrichment analysis of the PPI network of the top up-regulated protein-coding genes in ETMR identified their involvement in post-transcriptional regulatory pathways like mRNA processing and splicing, protein ubiquitination, and autophagy. Interestingly, an emerging role of LIN28A in carcinogenesis has been identified as being a modulator of alternative splicing and gene expression through regulation and interactions with splicing regulators like HNRNPA1 [13,48]. Additionally, the exogenous LIN28A expression displayed widespread splicing changes utilizing splicing-sensitive microarrays [14]. In our study, LIN28A and splicing factors like PUF60, HNRNPC, HNRNPA1, and HNRNPL showed increased expression compared to fetal controls. Furthermore, AMBRA1 (the activating molecule in BECN1-regulated autophagy protein 1) was found to be up-regulated in ETMR. Autophagy-related genes like AMBRA1 were shown to play a role in cell survival and chemotherapy resistance [49]. Moreover, heat shock proteins (HSPs) like HSPA4 and HSPA8 were also among the most up-regulated genes in ETMR. HSPs were demonstrated to be involved in protein quality control, the ubiquitin–proteasome system (UPS), endoplasmic reticulum (ER) associated with degradation and autophagy [48]. In addition, it has been previously reported that HSPA4 and HSPA8 were associated with poor prognosis in cancer [50,51]. It was also found that UBB was the most up-regulated gene in ETMR compared to the controls. Tian and colleagues showed that increased expression of UBB was important for cancer initiation and maintenance of the cancer stem cell state [52]. Interestingly, the knockdown of UBB via small interfering RNA led to inhibition of survival and proliferation of tumor cells by suppressing ubiquitination at multiple sites associated with cancer pathways, and by impeding the ability of tumor cells to overcome increased stress [53].
The link between aberrant AS and signaling pathways in cancer has been discussed before [20,54,55]. In our study, DEGs with altered splicing events in ETMR were enriched in survival signaling pathways such as mTOR, MAPK pathways as well as ubiquitin-mediated proteolysis and cell cycle. A previous study also demonstrated the involvement of overexpression of LIN28A in the activation of the mTOR pathway indicating that inhibitors targeting the IGF/PI3K/mTOR pathway could be a promising novel treatment of ETMR [47].
Nuclear export was among the biological processes enriched by the up-regulated having significant AS events. Interestingly, the exportin-1 (XPO1) gene was among the most up-regulated genes and displayed retained intron events in ETMR. XPO1 is one of the key mediators of nuclear export, which is a crucial step in intracellular signaling, and it is utilized by cancer cells to stimulate cell proliferation and evade apoptosis [56]. XPO1 was found to be up-regulated in several cancer types and was demonstrated to dysregulate intracellular localization of tumor suppressors and oncogenes, modulate autophagy, and contribute to tumor growth and progression [57,58]. Therefore, nuclear export inhibition is considered a potential prognostic marker and therapeutic target for cancer [59,60,61]. Hence, further studies should be executed to detect the presence of the XPO1 protein in ETMR and to investigate the effect of the retained intron on its functions.
Finally, our efforts in this in silico study identified genes with altered AS events and their functional significance in ETMR and provided targets for the critical experimental validation. Due to the scarcity of ETMR cases, the sample size in our study was limited, and therefore, we recommend the establishment of multicenter-based studies for the experimental validation of this findings.
5. Conclusions
In conclusion, our study describes a potential underlying mechanism in the carcinogenesis of ETMR, which is altered AS. Further investigations are needed to validate the ETMR-specific splicing events, which can be a potential therapeutic target for this fatal embryonic tumor.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/11/9/1108/s1, Table S1: DEGs and enrichment analysis. Table S2: Enrichment analysis of the genes in the PPI. Table S3: Genes with AS events and enrichment analysis of DEGs with AS events. Table S4: Up-regulated genes with RI events and their enrichment analysis. Figure S1: Principal component analysis (PCA) of the ETMR and fetal control samples. Figure S2: Heatmap-based hierarchical clustering of the samples.
Author Contributions
D.H.; methodology, data curation, S.E.-N.; supervision, D.H. and S.E.-N.; writing—original draft preparation, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Pickles, J.C.; Hawkins, C.; Pietsch, T.; Jacques, T.S. CNS embryonal tumours: WHO 2016 and beyond. Neuropathol. Appl. Neurobiol. 2018, 44, 151–162. [Google Scholar] [CrossRef]
- Korshunov, A.; Sturm, D.; Ryzhova, M.; Hovestadt, V.; Gessi, M.; Jones, D.T.W.; Remke, M.; Northcott, P.; Perry, A.; Picard, D.; et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol. 2014, 128, 279–289. [Google Scholar] [CrossRef]
- Spence, T.; Sin-Chan, P.; Picard, D.; Barszczyk, M.; Hoss, K.; Lu, M.; Kim, S.K.; Ra, Y.S.; Nakamura, H.; Fangusaro, J.; et al. CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol. 2014, 128, 291–303. [Google Scholar] [CrossRef]
- Geyer, J.R.; Sposto, R.; Jennings, M.; Boyett, J.M.; Axtell, R.A.; Breiger, D.; Broxson, E.; Donahue, B.; Finlay, J.L.; Goldwein, J.W.; et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: A Report from the Children’s Cancer Group. J. Clin. Oncol. 2005, 23, 7621–7631. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, S.H.; Shim, K.W.; Han, J.W.; Choi, J.; Kim, D.S.; Lyu, C.J.; Kim, J.W.; Suh, C.O.; Cho, J. Treatment outcome and prognostic molecular markers of supratentorial primitive neuroectodermal tumors. PLoS ONE 2016, 11, e0153443. [Google Scholar] [CrossRef]
- Picard, D.; Miller, S.; Hawkins, C.E.; Bouffet, E.; Rogers, H.A.; Chan, T.S.Y.; Kim, S.K.; Ra, Y.S.; Fangusaro, J.; Korshunov, A.; et al. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: An integrative genomic analysis. Lancet Oncol. 2012, 13, 838–848. [Google Scholar] [CrossRef]
- Gessi, M.; Giangaspero, F.; Lauriola, L.; Gardiman, M.; Scheithauer, B.W.; Halliday, W.; Hawkins, C.; Rosenblum, M.K.; Burger, P.C.; Eberhart, C.G. Embryonal tumors with abundant neuropil and true Rosettes: A distinctive CNS primitive neuroectodermal tumor. Am. J. Surg. Pathol. 2009, 33, 211. [Google Scholar] [CrossRef]
- Eberhart, C.G.; Brat, D.J.; Cohen, K.J.; Burger, P.C. Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr. Dev. Pathol. 2000, 3, 346–352. [Google Scholar] [CrossRef]
- Li, M.; Lee, K.F.; Lu, Y.; Clarke, I.; Shih, D.; Eberhart, C.; Collins, V.P.; Van Meter, T.; Picard, D.; Zhou, L.; et al. Frequent Amplification of a chr19q13.41 MicroRNA Polycistron in Aggressive Primitive Neuroectodermal Brain Tumors. Cancer Cell 2009, 16, 533–546. [Google Scholar] [CrossRef]
- Kleinman, C.L.; Gerges, N.; Papillon-Cavanagh, S.; Sin-Chan, P.; Pramatarova, A.; Quang, D.A.K.; Adoue, V.; Busche, S.; Caron, M.; Djambazian, H.; et al. Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat. Genet. 2014, 46, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843. [Google Scholar] [CrossRef]
- Yang, J.; Bennett, B.D.; Luo, S.; Inoue, K.; Grimm, S.A.; Schroth, G.P.; Bushel, P.R.; Kinyamu, H.K.; Archer, T.K. LIN28A modulates splicing and gene expression programs in breast cancer cells. Mol. Cell. Biol. 2015, 35, 3225–3243. [Google Scholar] [CrossRef]
- Wilbert, M.L.; Huelga, S.C.; Kapeli, K.; Stark, T.J.; Liang, T.Y.; Chen, S.X.; Yan, B.Y.; Nathanson, J.L.; Hutt, K.R.; Lovci, M.T.; et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol. Cell 2012, 48, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, N.; Lu, Z.; Sun, S.; Huang, J.; Chen, Z.; He, J. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. 2017, 393, 40–51. [Google Scholar] [CrossRef]
- Ladomery, M. Aberrant alternative splicing is another hallmark of cancer. Int. J. Cell Biol. 2013, 2013, 463786. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P. Unbalanced alternative splicing and its significance in cancer. BioEssays 2006, 28, 378–386. [Google Scholar] [CrossRef]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative Splicing and Tumor Progression. Curr. Genom. 2008, 9, 556–570. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hu, Z.; Zhao, Y.; Huang, S.; He, X. Transcriptome-Wide Analysis Reveals the Landscape of Aberrant Alternative Splicing Events in Liver Cancer. Hepatology 2019, 69, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Vagner, S.; Auboeuf, D. Alternative splicing and breast cancer. RNA Biol. 2010, 7, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, S.; Gallo, S.; Catillo, M.; Pignataro, D.; Biamonti, G.; Ghigna, C. Oncogenic alternative splicing switches: Role in cancer progression and prospects for therapy. Int. J. Cell Biol. 2013, 2013, 962038. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Yocum, A.K.; Menon, R. Alternative splice variants, a new class of protein cancer biomarker candidates: Findings in pancreatic cancer and breast cancer with systems biology implications. Dis. Markers 2010, 28, 241–251. [Google Scholar] [CrossRef]
- Samur, A.A.; Samur, M.K.; Lopez, M.A.; Derebail, S.; Anderson, K.C.; Avet-Loiseau, H.; Parmigiani, G.; Fulciniti, M.; Munshi, N.C. Genome wide transcriptomic analysis identifies dysregulated splicing factor profile with molecular and functional role in multiple myeloma. Blood 2019, 134, 361. [Google Scholar] [CrossRef]
- Lindsay, S.J.; Xu, Y.; Lisgo, S.N.; Harkin, L.F.; Copp, A.J.; Gerrelli, D.; Clowry, G.J.; Talbot, A.; Keogh, M.J.; Coxhead, J.; et al. HDBR Expression: A Unique Resource for Global and Individual Gene Expression Studies during Early Human Brain Development. Front. Neuroanat. 2016, 10, 86. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. R Package Version 1.3.5. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 4 August 2020).
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Rätsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; Von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pagliarini, V.; Naro, C.; Sette, C. Splicing regulation: A molecular device to enhance cancer cell adaptation. BioMed Res. Int. 2015, 2015, 543067. [Google Scholar] [CrossRef]
- Wan, Q.; Sang, X.; Jin, L.; Wang, Z. Alternative splicing events as indicators for the prognosis of uveal melanoma. Genes 2020, 11, 227. [Google Scholar] [CrossRef]
- Xue, D.; Cheng, P.; Jiang, J.; Ren, Y.; Wu, D.; Chen, W. Systemic Analysis of the Prognosis-Related RNA Alternative Splicing Signals in Melanoma. Med. Sci. Monit. 2020, 26, e921133-1. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, Q.; Liu, X.; Ji, Y.; Chao, H.-P.; Liu, Y.; Tracz, A.; Kirk, J.; Buonamici, S.; Zhu, P.; et al. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat. Commun. 2020, 11, 2089. [Google Scholar] [CrossRef]
- Suzuki, H.; Kumar, S.A.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; De Antonellis, P.; Cavalli, F.M.G.; Juraschka, K.; Farooq, H.; Shibahara, I.; et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef]
- Huang, H.H.; Ferguson, I.D.; Thornton, A.M.; Bastola, P.; Lam, C.; Lin, Y.H.T.; Choudhry, P.; Mariano, M.C.; Marcoulis, M.D.; Teo, C.F.; et al. Proteasome inhibitor-induced modulation reveals the spliceosome as a specific therapeutic vulnerability in multiple myeloma. Nat. Commun. 2020, 11, 1931. [Google Scholar] [CrossRef] [PubMed]
- Urbanski, L.M.; Leclair, N.; Anczuków, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Ryzhova, M.; Jones, D.T.W.; Northcott, P.A.; Van Sluis, P.; Volckmann, R.; Koster, J.; Versteeg, R.; Cowdrey, C.; Perry, A.; et al. LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol. 2012, 124, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Ng, S.C.; Segr, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef]
- Spence, T.; Perotti, C.; Sin-Chan, P.; Picard, D.; Wu, W.; Singh, A.; Anderson, C.; Blough, M.D.; Cairncross, J.G.; Lafay-Cousin, L.; et al. A novel C19MC amplified cell line links Lin28/let-7 to mTOR signaling in embryonal tumor with multilayered rosettes. Neuro. Oncol. 2014, 16, 62–71. [Google Scholar] [CrossRef]
- Xiong, H.; Zhao, W.; Wang, J.; Seifer, B.J.; Ye, C.; Chen, Y.; Jia, Y.; Chen, C.; Shen, J.; Wang, L.; et al. Oncogenic mechanisms of Lin28 in breast cancer: New functions and therapeutic opportunities. Oncotarget 2017, 8, 25721. [Google Scholar] [CrossRef]
- Sun, W.L. Ambra1 in autophagy and apoptosis: Implications for cell survival and chemotherapy resistance. Oncol. Lett. 2016, 12, 367–374. [Google Scholar] [CrossRef]
- Bozaykut, P.; Ozer, N.K.; Karademir, B. Regulation of protein turnover by heat shock proteins. Free Radic. Biol. Med. 2014, 77, 195–209. [Google Scholar] [CrossRef]
- Zoppino, F.C.M.; Guerrero-Gimenez, M.E.; Castro, G.N.; Ciocca, D.R. Comprehensive transcriptomic analysis of heat shock proteins in the molecular subtypes of human breast cancer. BMC Cancer 2018, 18, 700. [Google Scholar] [CrossRef]
- Tian, Y.; Ding, W.; Wang, Y.; Ji, T.; Sun, S.; Mo, Q.; Chen, P.; Fang, Y.; Liu, J.; Wang, B.; et al. Ubiquitin B in cervical cancer: Critical for the maintenance of cancer stem-like cell characters. PLoS ONE 2013, 8, e84457. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Park, S.; Lee, E.K.; Yoo, Y.J. Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci. Rep. 2013, 3, 2623. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.; Pereira, J.F.S.; Jordan, P. Signaling pathways driving aberrant splicing in cancer cells. Genes 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- El Marabti, E.; Younis, I. The cancer spliceome: Reprograming of alternative splicing in cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Tran, E.J.; King, M.C.; Corbett, A.H. Macromolecular transport between the nucleus and the cytoplasm: Advances in mechanism and emerging links to disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 85. [Google Scholar] [CrossRef]
- Martin, A.P.; Jacquemyn, M.; Lipecka, J.; Chhuon, C.; Aushev, V.N.; Meunier, B.; Singh, M.K.; Carpi, N.; Piel, M.; Codogno, P.; et al. STK 38 kinase acts as XPO 1 gatekeeper regulating the nuclear export of autophagy proteins and other cargoes. EMBO Rep. 2019, 20, e48150. [Google Scholar] [CrossRef]
- Niu, M.; Wu, S.; Mao, L.; Yang, Y. CRM1 is a cellular target of curcumin: New insights for the myriad of biological effects of an ancient spice. Traffic 2013, 14, 1042–1052. [Google Scholar] [CrossRef]
- Yashiroda, Y.; Yoshida, M. Nucleo-Cytoplasmic Transport of Proteins as a Target for Therapeutic Drugs. Curr. Med. Chem. 2005, 10, 741–748. [Google Scholar] [CrossRef]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (SINE)-A novel class of anti-cancer agents. J. Hematol. Oncol. 2014, 7, 78. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).