Beyond Back Splicing, a Still Poorly Explored World: Non-Canonical Circular RNAs



{kind=link}
{kind=link}
Abstract
:1. Introduction
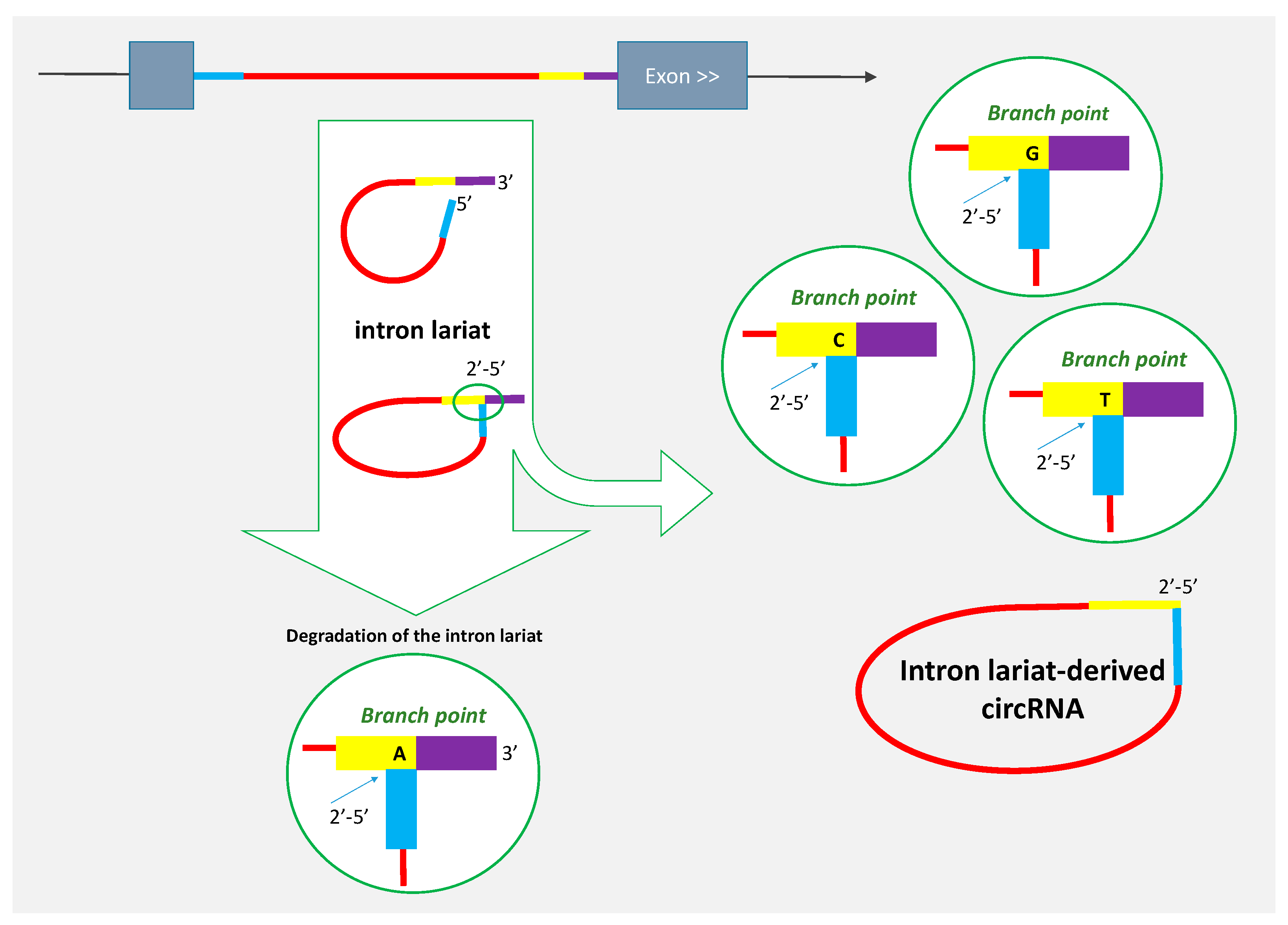
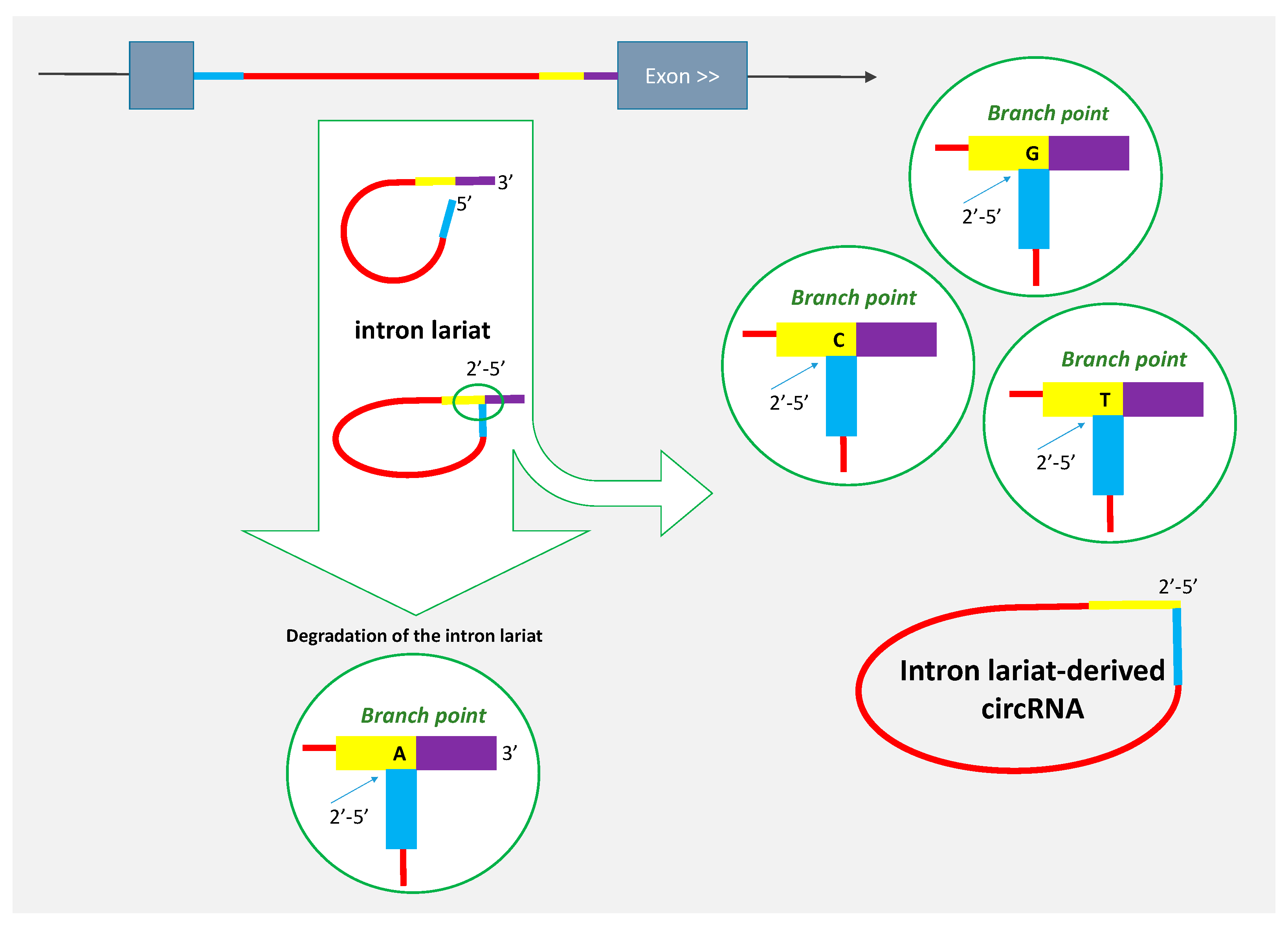
2. Some circRNAs Are Derived from Intron Lariats
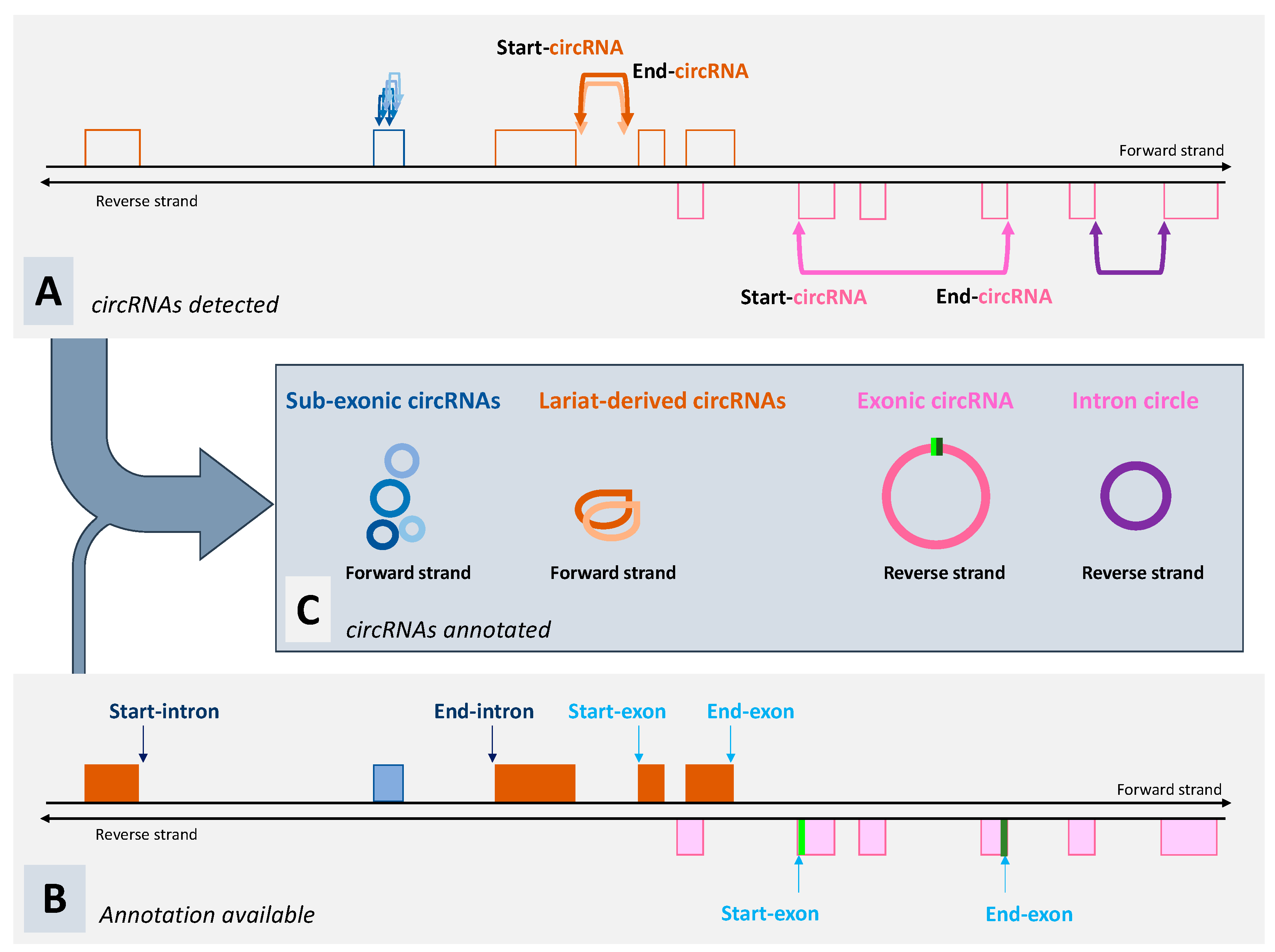
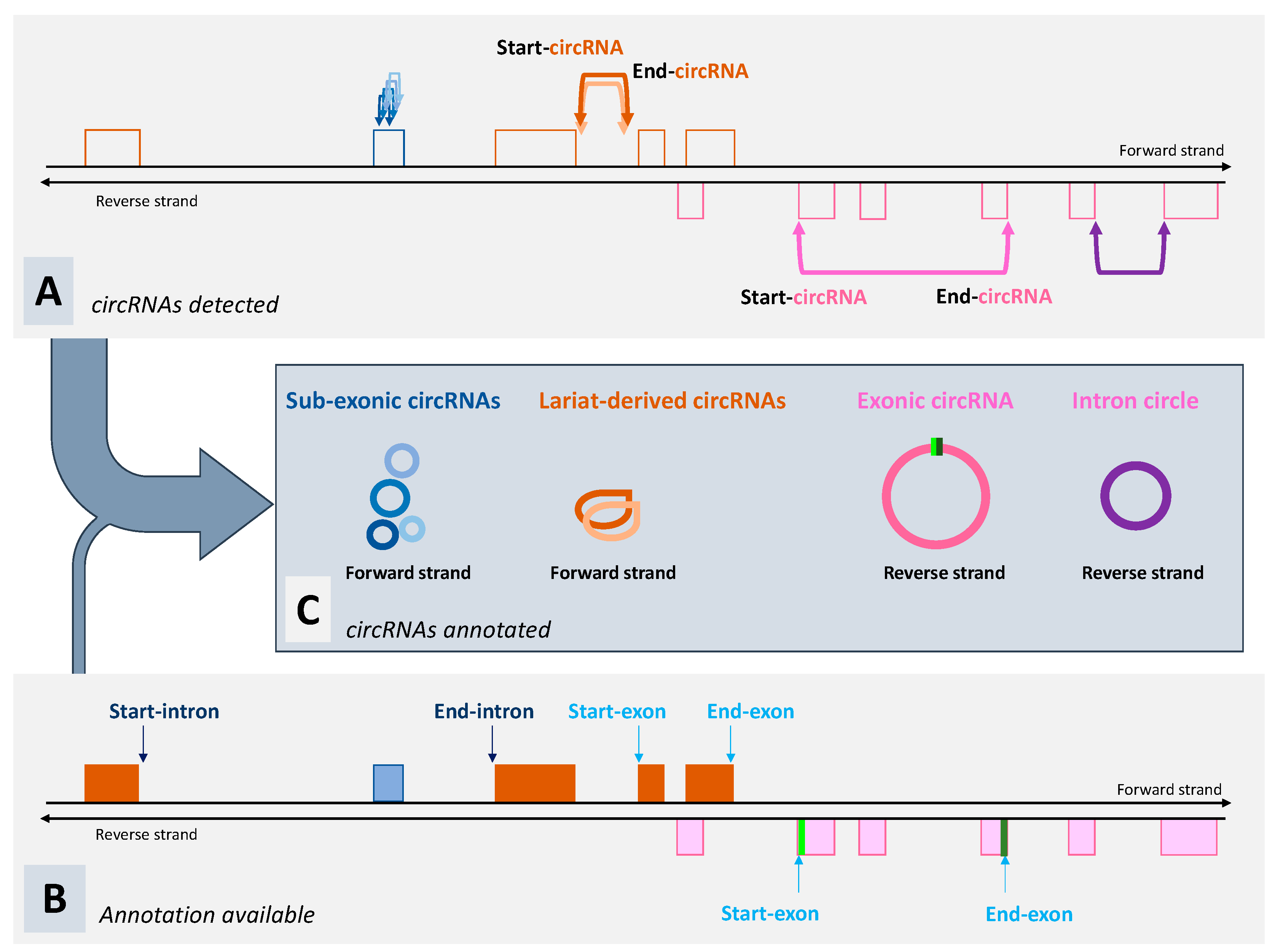
3. What Can We Expect from Tools Dedicated to the Detection of circRNA?
4. Review of Reported Lariat-Derived circRNA to Underline Features Related to Their Genesis
5. Why Have Lariat-Derived Intronic circRNAs Not Been Better Investigated?
6. Non-Canonical circRNAs Are Even More Poorly Known Than Lariat-Derived circRNAs
7. What Is the Function of Non-Canonical circRNA?
8. How Can We Improve Our Knowledge of Non-Canonical circRNAs?
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, W. The RNA lariat: A new ring to the splicing of mRNA precursors. Cell 1984, 39, 423–425. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Zhang, L.; Issaian, A.; Hill, R.C.; Espinosa, S.; Shi, S.; Cui, Y.; Kappel, K.; Das, R.; et al. A unified mechanism for intron and exon definition and back-splicing. Nature 2019, 573, 375–380. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Chen, L.L. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [Green Version]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Xiao, M.S.; Ai, Y.; Wilusz, J.E. Biogenesis and Functions of Circular RNAs Come into Focus. Trends Cell Biol. 2020, 30, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Ma, X.L.; Li, H.G. Intriguing circles: Conflicts and controversies in circular RNA research. RNA 2019, 10, e1538. [Google Scholar] [CrossRef] [PubMed]
- Patop, I.L.; Wust, S.; Kadener, S. Past, present, and future of circRNAs. EMBO J. 2019, 38, e100836. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, A.; Rosbash, M. RNA splicing and intron turnover are greatly diminished by a mutant yeast branch point. Proc. Natl. Acad. Sci. USA 1986, 83, 5835–5839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Clark, M.B.; Andersen, S.B.; Brunck, M.E.; Haerty, W.; Crawford, J.; Taft, R.J.; Nielsen, L.K.; Dinger, M.E.; Mattick, J.S. Genome-wide discovery of human splicing branchpoints. Genome Res. 2015, 25, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, E.J.; Nizami, Z.F.; Talbot, C.C., Jr.; Gall, J.G. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev. 2012, 26, 2550–2559. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.N.; Pek, J.W. Stable Intronic Sequence RNAs (sisRNAs): An Expanding Universe. Trends Biochem. Sci. 2019, 44, 258–272. [Google Scholar] [CrossRef]
- Osman, I.; Tay, M.L.; Pek, J.W. Stable intronic sequence RNAs (sisRNAs): A new layer of gene regulation. Cell Mol. Life Sci. 2016, 73, 3507–3519. [Google Scholar] [CrossRef]
- Robic, A.; Demars, J.; Kühn, C. In-depth analysis reveals production of circular RNAs from non-coding sequences. Cells 2020, 9, 1806. [Google Scholar] [CrossRef]
- Robic, A.; Faraut, T.; Djebali, S.; Weikard, R.; Feve, K.; Maman, S.; Kuehn, C. Analysis of pig transcriptomes suggests a global regulation mechanism enabling temporary bursts of circular RNAs. RNA Biol. 2019, 16, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.; Bicknell, A.A.; Eddy, S.R.; Moore, M.J. Free circular introns with an unusual branchpoint in neuronal projections. Elife 2019, 8, e47809. [Google Scholar] [CrossRef] [PubMed]
- Talhouarne, G.J.S.; Gall, J.G. Lariat intronic RNAs in the cytoplasm of vertebrate cells. Proc. Natl. Acad. Sci. USA 2018, 115, E7970–E7977. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Das, A.; Sahu, M.; Mishra, S.S.; Khan, S.; Bejugam, P.R.; Rout, P.K.; Das, A.; Bano, S.; Mishra, G.P.; et al. Identification and Characterization of Circular Intronic RNAs Derived from Insulin Gene. Int. J. Mol. Sci. 2020, 21, 4302. [Google Scholar] [CrossRef] [PubMed]
- Talhouarne, G.J.; Gall, J.G. Lariat intronic RNAs in the cytoplasm of Xenopus tropicalis oocytes. RNA 2014, 20, 1476–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, C.; Sun, H.; Wang, J.; Liang, Y.; Wang, Y.; Wong, G. The bioinformatics toolbox for circRNA discovery and analysis. Brief Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhao, F. Computational Strategies for Exploring Circular RNAs. Trends Genet. 2018, 34, 389–400. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Kaur, S.; Mirza, A.H.; Pociot, F. Cell Type-Selective Expression of Circular RNAs in Human Pancreatic Islets. Noncoding RNA 2018, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Ragan, C.; Goodall, G.J.; Shirokikh, N.E.; Preiss, T. Insights into the biogenesis and potential functions of exonic circular RNA. Sci. Rep. 2019, 9, 2048. [Google Scholar] [CrossRef]
- Guo, Z.; Cao, Q.; Zhao, Z.; Song, C. Biogenesis, Features, Functions, and Disease Relationships of a Specific Circular RNA: CDR1as. Aging Dis. 2020, 11, 1009–1020. [Google Scholar] [CrossRef]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011, 30, 4414–4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, S.P.; Parker, K.R.; Horn, C.; Mata, M.; Salzman, J. ciRS-7 exonic sequence is embedded in a long non-coding RNA locus. PLoS Genet. 2017, 13, e1007114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.O.; Dong, R.; Zhang, Y.; Zhang, J.L.; Luo, Z.; Zhang, J.; Chen, L.L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Taggart, A.J.; Lin, C.L.; Shrestha, B.; Heintzelman, C.; Kim, S.; Fairbrother, W.G. Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 2017, 27, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; He, X.; Silva, E. Stable intronic sequence RNAs (sisRNAs) are selected regions in introns with distinct properties. BMC Genomics 2020, 21, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268, corrected in J. Appl. Genet. 2019, 60, 231. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Kameyama, T.; Ohe, K.; Tsukahara, T.; Mayeda, A. Nested introns in an intron: Evidence of multi-step splicing in a large intron of the human dystrophin pre-mRNA. FEBS Lett. 2013, 587, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Lorsch, J.R.; Bartel, D.P.; Szostak, J.W. Reverse transcriptase reads through a 2′-5′linkage and a 2′-thiophosphate in a template. Nucleic Acids Res. 1995, 23, 2811–2814. [Google Scholar] [CrossRef]
- Das, A.; Rout, P.K.; Gorospe, M.; Panda, A.C. Rolling Circle cDNA Synthesis Uncovers Circular RNA Splice Variants. Int. J. Mol. Sci. 2019, 20, 3988. [Google Scholar] [CrossRef] [Green Version]
- Wan, R.; Bai, R.; Zhan, X.; Shi, Y. How Is Precursor Messenger RNA Spliced by the Spliceosome? Annu. Rev. Biochem. 2020, 89, 333–358. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, Z.; Zhou, J.; Tian, C.; Tian, G.; He, M.; Gao, L.; Chen, L.; Li, T.; Peng, H.; et al. Interior circular RNA. RNA Biol. 2020, 17, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Danan, M.; Schwartz, S.; Edelheit, S.; Sorek, R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012, 40, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Filonov, G.S.; Noto, J.J.; Schmidt, C.A.; Hatkevich, T.L.; Wen, Y.; Jaffrey, S.R.; Matera, A.G. Metazoan tRNA introns generate stable circular RNAs in vivo. RNA 2015, 21, 1554–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.A.; Giusto, J.D.; Bao, A.; Hopper, A.K.; Matera, A.G. Molecular determinants of metazoan tricRNA biogenesis. Nucleic Acids Res. 2019, 47, 6452–6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervera, A.; de la Pena, M. Small circRNAs with self-cleaving ribozymes are highly expressed in diverse metazoan transcriptomes. Nucleic Acids Res. 2020, 48, 5054–5064. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Zuo, Y.; Wang, J.; Zhang, M.Q.; Malhotra, A.; Mayeda, A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006, 34, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susuki, H.; Tsukahara, T. A View of Pre-mRNA Splicing from RNase R Resistant RNAs. Int. J. Mol. Sci. 2014, 15, 9331–9342. [Google Scholar] [CrossRef] [Green Version]
- Boivin, V.; Reulet, G.; Boisvert, O.; Couture, S.; Elela, S.A.; Scott, M.S. Reducing the structure bias of RNA-Seq reveals a large number of non-annotated non-coding RNA. Nucleic Acids Res. 2020, 48, 2271–2286. [Google Scholar] [CrossRef]
- Dahl, M.; Daugaard, I.; Andersen, M.S.; Hansen, T.B.; Gronbaek, K.; Kjems, J.; Kristensen, L.S. Enzyme-free digital counting of endogenous circular RNA molecules in B-cell malignancies. Lab. Investig. 2018, 98, 1657–1669. [Google Scholar] [CrossRef] [Green Version]
- Conn, V.M.; Conn, S.J. SplintQuant: A method for accurately quantifying circular RNA transcript abundance without reverse transcription bias. RNA 2019, 25, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robic, A.; Kühn, C. Beyond Back Splicing, a Still Poorly Explored World: Non-Canonical Circular RNAs. Genes 2020, 11, 1111. https://doi.org/10.3390/genes11091111
Robic A, Kühn C. Beyond Back Splicing, a Still Poorly Explored World: Non-Canonical Circular RNAs. Genes. 2020; 11(9):1111. https://doi.org/10.3390/genes11091111
Chicago/Turabian StyleRobic, Annie, and Christa Kühn. 2020. "Beyond Back Splicing, a Still Poorly Explored World: Non-Canonical Circular RNAs" Genes 11, no. 9: 1111. https://doi.org/10.3390/genes11091111
APA StyleRobic, A., & Kühn, C. (2020). Beyond Back Splicing, a Still Poorly Explored World: Non-Canonical Circular RNAs. Genes, 11(9), 1111. https://doi.org/10.3390/genes11091111