Inhibition of miRNA-34a Promotes M2 Macrophage Polarization and Improves LPS-Induced Lung Injury by Targeting Klf4

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Differentially Expressed Genes (DEGs) Screening from the mRNA Expression Profile
2.2. Significant miRNAs and Transcription Factors (TFs) Retrieval for Feed-Forward Loop (FFL) Construction
2.3. Materials
2.4. Murine ALI Model
2.5. Cell Culture and Transient Transfection
2.6. Macrophage Polarization
2.7. Histopathological Analyses
2.8. Semiquantitative PCR
2.9. miRNA Quantification
2.10. Western Blot Analysis
2.11. Statistical Analyses
3. Results
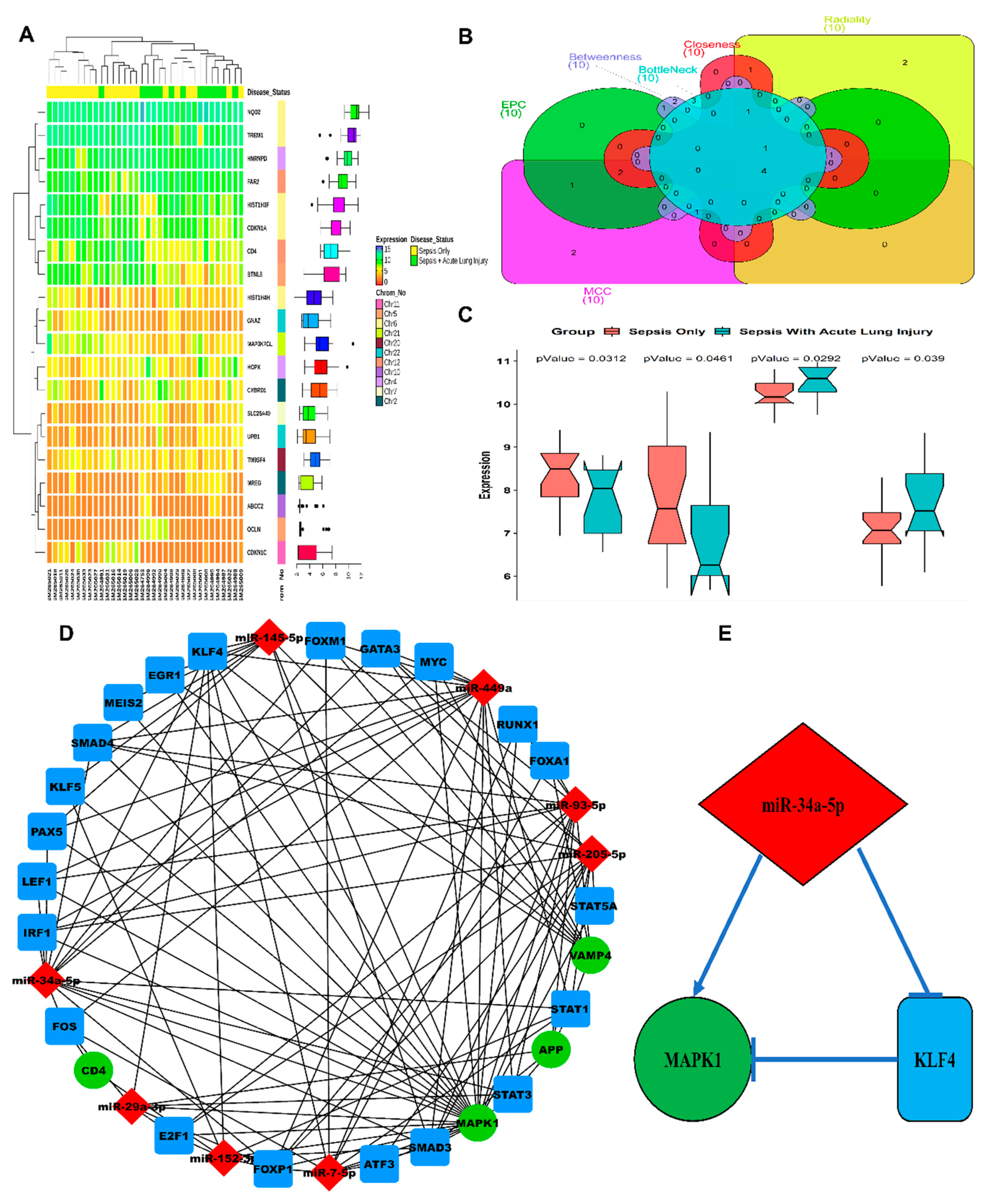
3.1. ALI-Associated DEGs, miRNAs, and Hub Gene(s) Prediction
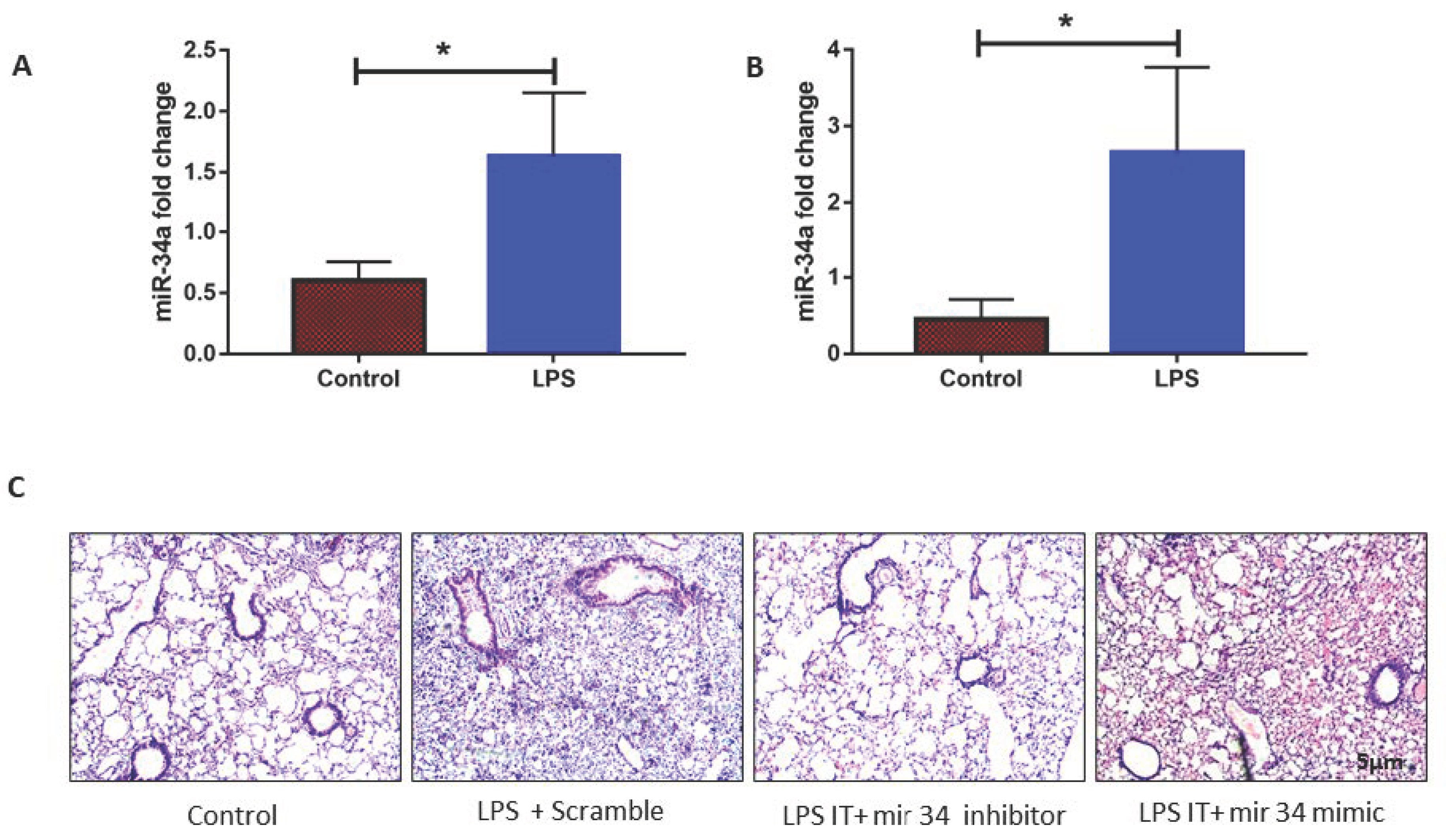
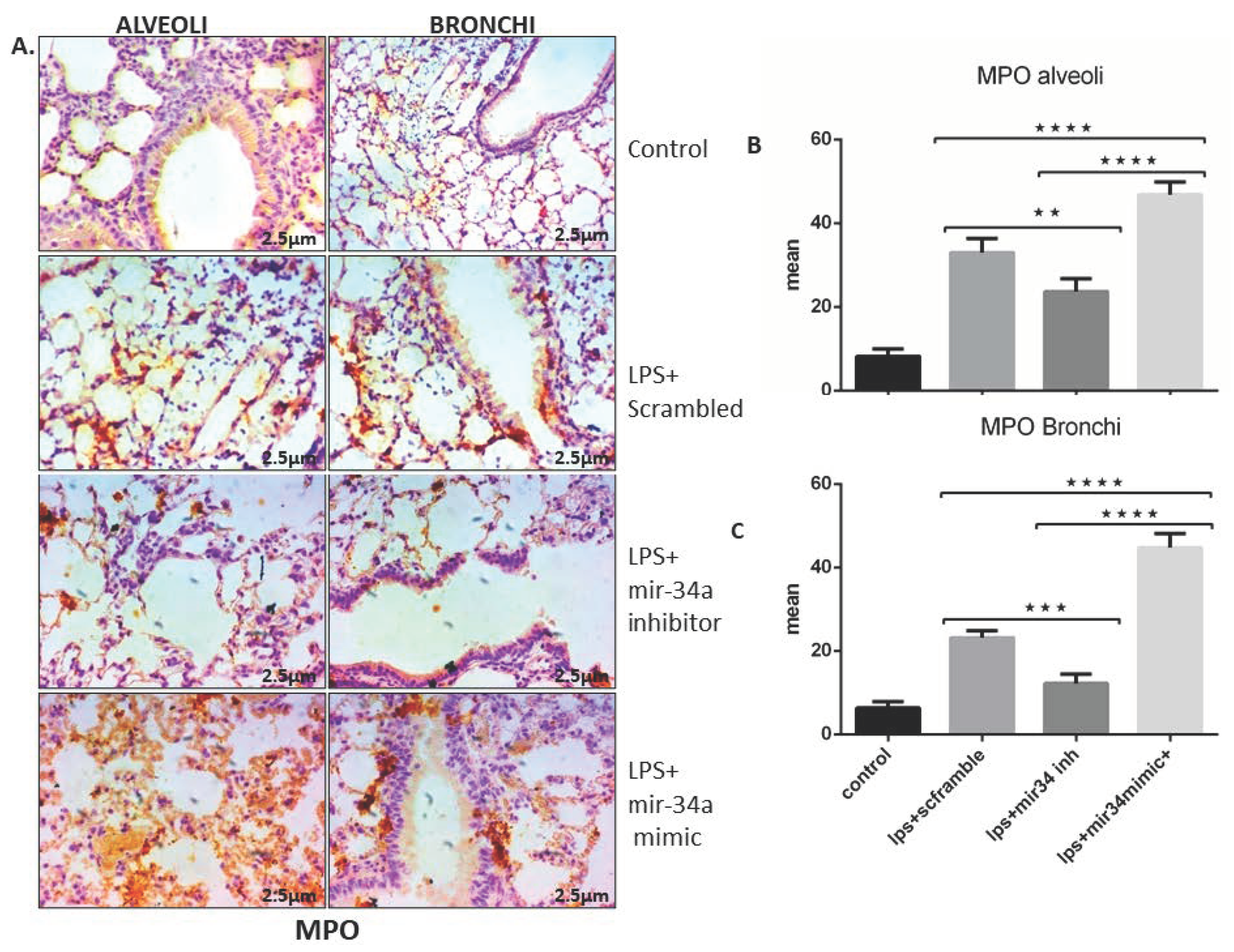
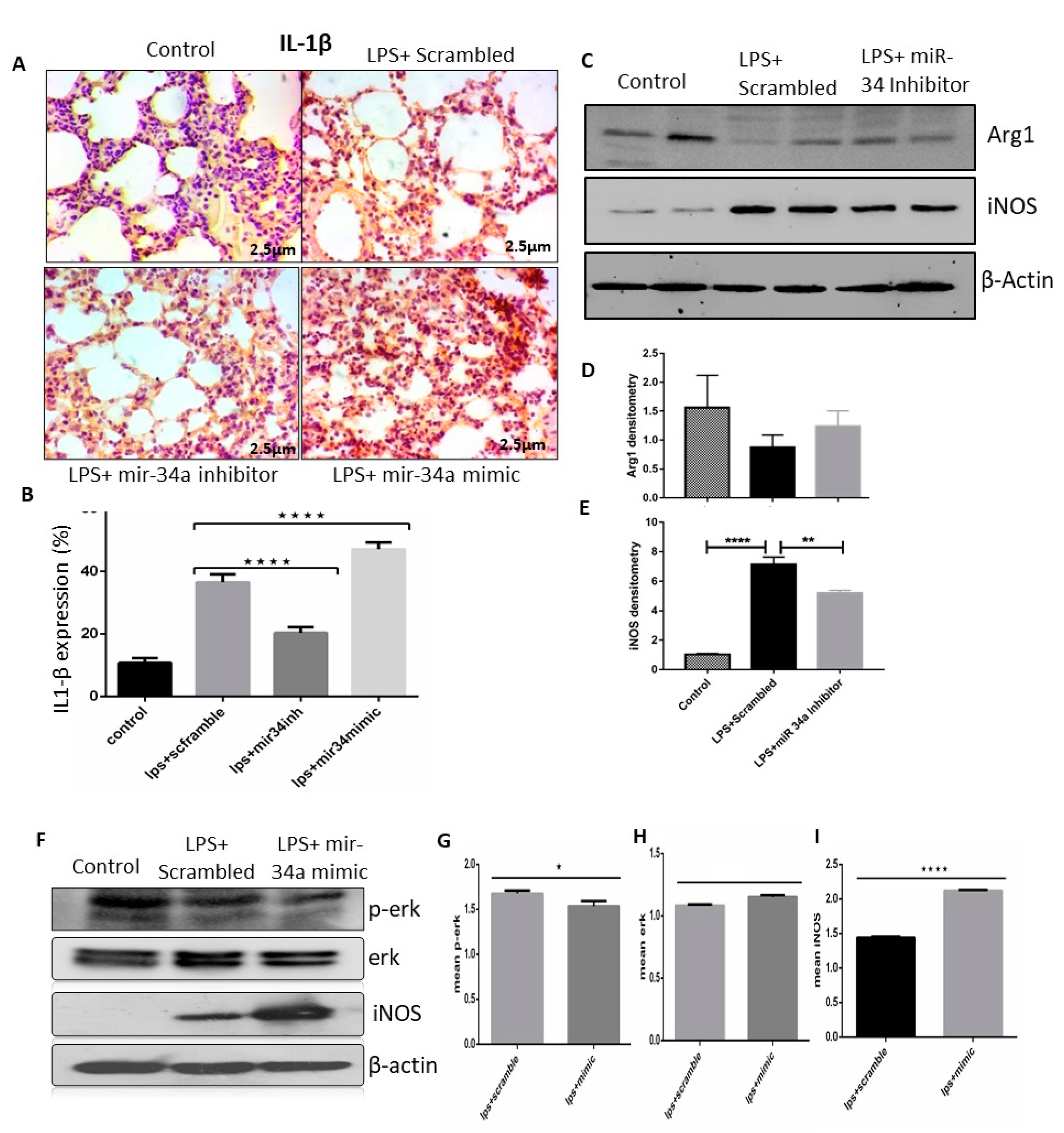
3.2. LPS Induced mir-34a in Lung Macrophages Accompanied with the Severity of Lung Injury
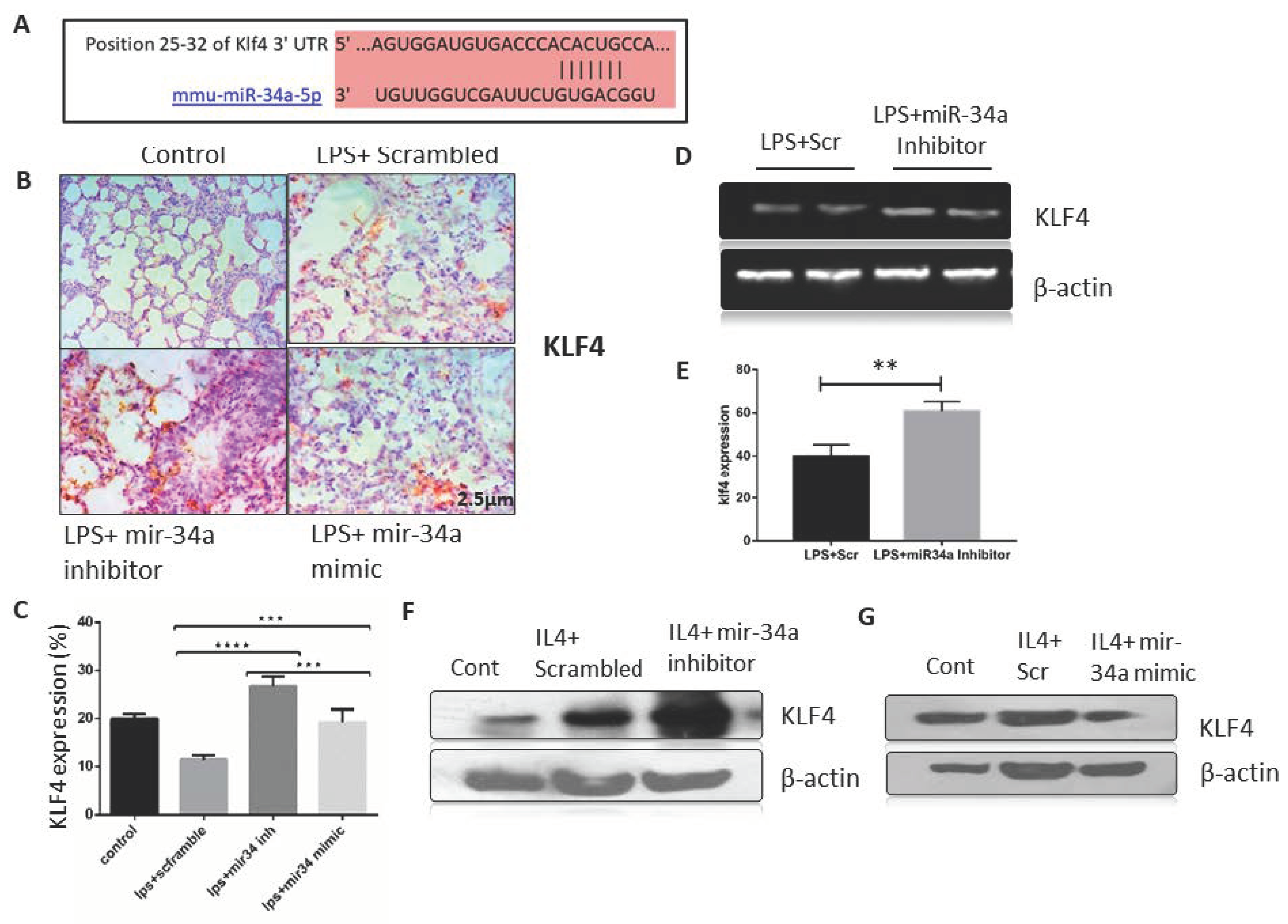
3.3. miR-34a Expression Downregulates Klf4 Expression in Macrophages
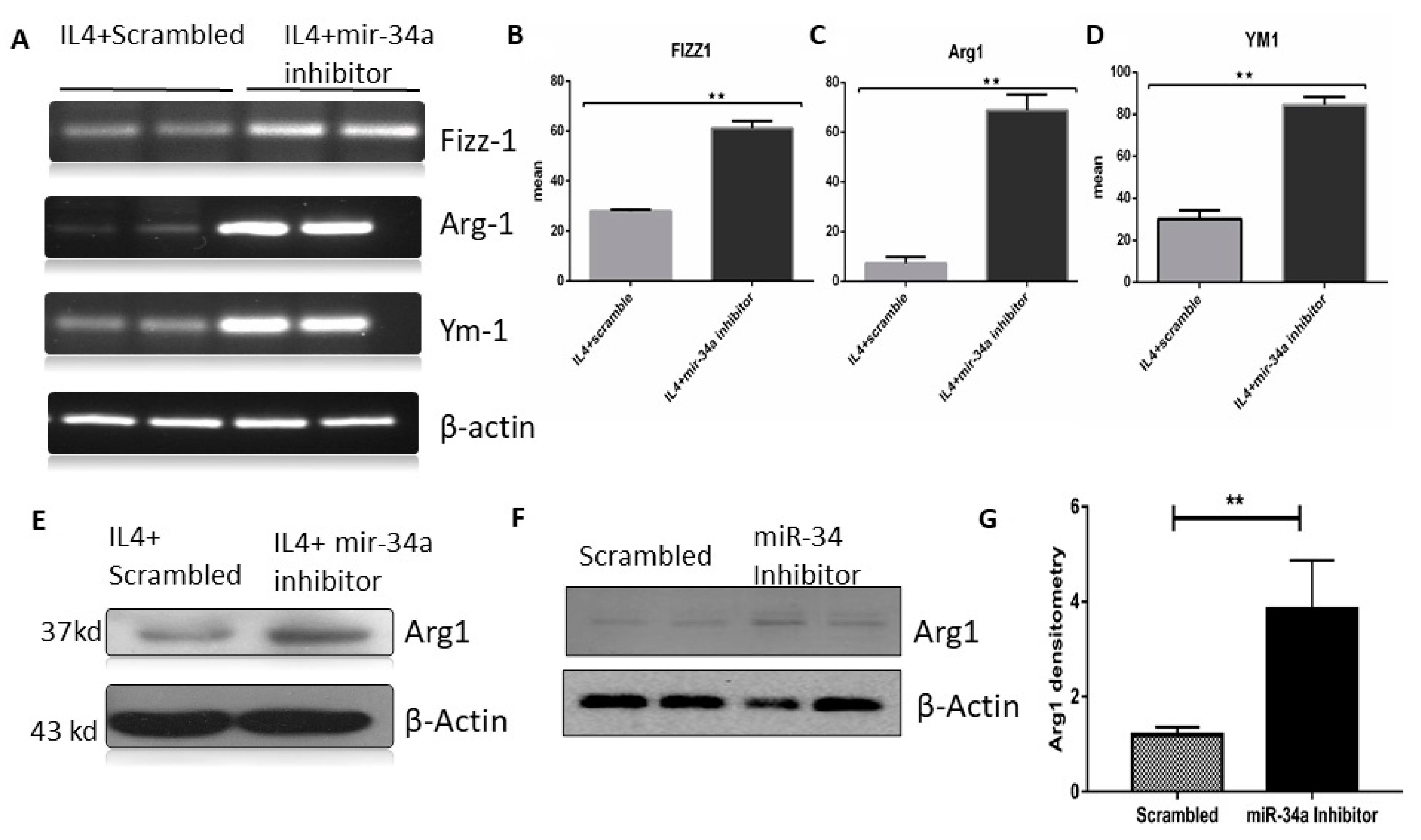
3.4. miR-34a Regulates Macrophage Polarization
3.5. miR-34a Augments the Effects of LPS Arresting Cell Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; LeGall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Panda, S.K.; Agarwal, B.; Behera, S.; Ali, S.M.; Pulse, M.E.; Solomkin, J.S.; Opal, S.M.; Bhandari, V.; Acharya, S. Novel chitohexaose analog protects young and aged mice from CLP Induced polymicrobial sepsis. Sci. Rep. 2019, 9, 2904. [Google Scholar] [CrossRef] [PubMed]
- Umbrello, M.; Formenti, P.; Bolgiaghi, L.; Chiumello, D. Current concepts of ARDS: A narrative review. Int. J. Mol. Sci. 2016, 18, 64. [Google Scholar] [CrossRef] [Green Version]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 2019, 5, 18. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Corrigendum: Macrophage polarization: Different gene signatures in M1(LPS+) vs. classically and M2(LPS–) vs. alternatively activated macrophages. Front. Immunol. 2020, 11, 234. [Google Scholar] [CrossRef]
- Herold, S.; Mayer, K.; Lohmeyer, J. Acute lung injury: How macrophages orchestrate resolution of inflammation and tissue repair. Front. Immunol. 2011, 2, 65. [Google Scholar] [CrossRef] [Green Version]
- Benoit, M.; Desnues, B.; Mege, J.-L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Liu, G.; Abraham, E. MicroRNAs in immune response and macrophage polarization. Arter. Thromb. Vasc. Boil. 2013, 33, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.A.; Bhandari, V. Hyperoxia exacerbates postnatal inflammation-induced lung injury in neonatal BRP-39 null mutant mice promoting the M1 macrophage phenotype. Mediat. Inflamm. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2017, 223, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M.A.; Arora, S.; Prakasam, G.; Calin, G.A.; Syed, M. MicroRNA in lung cancer: Role, mechanisms, pathways and therapeutic relevance. Mol. Asp. Med. 2019, 70, 3–20. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Tasena, H.; Faiz, A.; Timens, W.; Noordhoek, J.; Hylkema, M.N.; Gosens, R.; Hiemstra, P.S.; Spira, A.; Postma, D.S.; Tew, G.W.; et al. microRNA–mRNA regulatory networks underlying chronic mucus hypersecretion in COPD. Eur. Respir. J. 2018, 52, 1701556. [Google Scholar] [CrossRef]
- Lu, X.-G.; Kang, X.; Zhan, L.-B.; Kang, L.-M.; Fan, Z.-W.; Bai, L.-Z. Circulating miRNAs as biomarkers for severe acute pancreatitis associated with acute lung injury. World J. Gastroenterol. 2017, 23, 7440–7449. [Google Scholar] [CrossRef]
- Lee, W.; Kim, I.; Shin, S.; Park, K.; Yang, K.; Eun, J.W.; Sul, H.; Jeong, S. Expression profiling of microRNAs in lipopolysaccharide-induced acute lung injury after hypothermia treatment. Mol. Cell. Toxicol. 2016, 12, 243–253. [Google Scholar] [CrossRef]
- Bartel, S.; Schulz, N.; Alessandrini, F.; Schamberger, A.C.; Pagel, P.; Theis, F.J.; Milger, K.; Noessner, E.; Stick, S.; Kicic, A.; et al. Pulmonary microRNA profiles identify involvement of Creb1 and Sec14l3 in bronchial epithelial changes in allergic asthma. Sci. Rep. 2017, 7, 46026. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Shields, J.M.; Christy, R.J.; Yang, V.W. Identification and characterization of a gene encoding a gut-enriched Krüppel-like factor expressed during growth arrest. J. Boil. Chem. 1996, 271, 20009–20017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, M.W.; Wara, A.K.; Cao, Z.; Lebedeva, M.A.; Rosenbauer, F.; Iwasaki, H.; Hirai, H.; Katz, J.P.; Haspel, R.L.; Gray, S.; et al. The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J. 2007, 26, 4138–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flandez, M.; Guilmeau, S.; Blache, P.; Augenlicht, L. KLF4 regulation in intestinal epithelial cell maturation. Exp. Cell Res. 2008, 314, 3712–3723. [Google Scholar] [CrossRef] [Green Version]
- Cowan, C.E.; Kohler, E.E.; Dugan, T.A.; Mirza, M.K.; Malik, A.B.; Wary, K.K. Kruppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. Circ. Res. 2010, 107, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.-L.; Fang, H.-X.; Liang, Y.; Zhao, Y.; Shi, C.-S. MicroRNA-34a promotes iNOS secretion from pulmonary macrophages in septic suckling rats through activating STAT3 pathway. Biomed. Pharmacother. 2018, 105, 1276–1282. [Google Scholar] [CrossRef]
- Das, P.; Curstedt, T.; Agarwal, B.; Prahaladan, V.M.; Ramirez, J.; Bhandari, S.; Syed, M.; Salomone, F.; Casiraghi, C.; Pelizzi, N.; et al. Small molecule inhibitor adjuvant surfactant therapy attenuates ventilator and hyperoxia-induced lung injury in preterm rabbits. Front. Physiol. 2020, 11, 266. [Google Scholar] [CrossRef]
- Das, P.; Syed, M.A.; Shah, D.; Bhandari, V. MiR34a: A master regulator in the pathogenesis of bronchopulmonary dysplasia. Cell Stress 2018, 2, 34–36. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Huang, M.; Ma, Y.-F. The effects of microRNA-34a regulating Notch-1/NF-κB signaling pathway on lipopolysaccharide-induced human umbilical vein endothelial cells. World J. Emerg. Med. 2017, 8, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Singh, P.; Sharma, A.; Arora, S.; Shriwash, N.; Rahmani, A.H.; Almatroodi, S.A.; Manda, K.; Dohare, R.; Syed, M.A. Transcriptome meta-analysis deciphers a dysregulation in immune response-associated gene signatures during sepsis. Genes 2019, 10, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Bruggen, T.; Nijenhuis, S.; Van Raaij, E.; Verhoef, J.; Van Asbeck, B.S. Lipopolysaccharide-induced tumor necrosis factor alpha production by human monocytes involves the Raf-1/MEK1-MEK2/ERK1-ERK2 pathway. Infect. Immun. 1999, 67, 3824–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Ahmad, S.; Irshad, R.; Goyal, Y.; Rafat, S.; Siddiqui, N.; Dev, K.; Husain, M.; Ali, S.; Mohan, A.; et al. TLRs in pulmonary diseases. Life Sci. 2019, 233, 116671. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Syed, M.A.; Panchal, D.; Joo, M.; Colonna, M.; Brantly, M.; Sadikot, R.T. Triggering receptor expressed on myeloid cells 1 (TREM-1)-mediated Bcl-2 induction prolongs macrophage survival. J. Boil. Chem. 2014, 289, 15118–15129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, K.; Pahl, A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochem. Pharmacol. 2009, 77, 1827–1834. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Hui, X.; Hoo, R.L.C.; Ye, D.-W.; Chan, C.Y.C.; Feng, T.; Wang, Y.; Lam, K.S.L.; Xu, A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Investig. 2019, 129, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.; Mohan, V.; Mansoor, S.; Tiruppathi, C.; Sadikot, R.T.; Natarajan, V. Role of nicotinamide adenine dinucleotide phosphate–reduced oxidase proteins in pseudomonas aeruginosa–induced lung inflammation and permeability. Am. J. Respir. Cell Mol. Boil. 2013, 48, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Clough, E.; Barrett, T. The gene expression omnibus database. Breast Cancer 2016, 1418, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhou, K.-R.; Liu, S.; Sun, W.-J.; Zheng, L.-L.; Zhou, H.; Yang, J.-H.; Qu, L.-H. ChIPBase v2.0: Decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2016, 45, D43–D50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.-C.; Ma’Ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. MiRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. StarBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2013, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173. [Google Scholar] [CrossRef]
- Ruiz-Camp, J.; Quantius, J.; Lignelli, E.; Arndt, P.F.; Palumbo, F.; Nardiello, C.; Solaligue, D.E.S.; Sakkas, E.; Mižíková, I.; Rodríguez-Castillo, J.A.; et al. Targeting miR-34a/ Pdgfra interactions partially corrects alveologenesis in experimental bronchopulmonary dysplasia. EMBO Mol. Med. 2019, 11, e9448. [Google Scholar] [CrossRef]
- Qin, L.-B.; Li, Z.-Y.; Li, H.; Fan, X.-Q.; Liu, H.-G.; Dong, X.-M.; Jia, W.-Y. Inhibitive effects of microRNA-34a on protecting against ischemia-reperfusion injury of vital organs in hemorrhagic shock pregnant mice. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1812–1818. [Google Scholar]
- Wang, K.; Zhou, W.; Cai, Q.; Cheng, J.; Cai, R.; Xing, R. SUMOylation of KLF4 promotes IL-4 induced macrophage M2 polarization. Cell Cycle 2017, 16, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, N.; Niu, J.; Saad, Y.; Kumar, S.; Sirakova, T.; Becerra, E.; Li, X.; Kolattukudy, P.E. Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J. Immunol. 2015, 194, 6011–6023. [Google Scholar] [CrossRef] [Green Version]
- Watters, J.J.; Sommer, J.A.; Pfeiffer, Z.A.; Prabhu, U.; Guerra, A.N.; Bertics, P.J. A differential role for the mitogen-activated protein kinases in lipopolysaccharide signaling. J. Boil. Chem. 2002, 277, 9077–9087. [Google Scholar] [CrossRef] [Green Version]
- Guha, M.; O’Connell, M.; Pawlinski, R.; Hollis, A.; McGovern, P.; Yan, S.-F.; Stern, D.; Mackman, N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor α expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 2001, 98, 1429–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghera, J.S.; Weinstein, S.L.; Aluwalia, M.; Girn, J.; Pelech, S.L. Activation of multiple proline-directed kinases by bacterial lipopolysaccharide in murine macrophages. J. Immunol. 1996, 156, 4457–4465. [Google Scholar]
- Yuan, Z.; Syed, M.; Panchal, D.; Joo, M.; Bedi, C.; Lim, S.; Önyüksel, H.; Rubinstein, I.; Colonna, M.; Sadikot, R.T. TREM-1-accentuated lung injury via miR-155 is inhibited by LP17 nanomedicine. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L426–L438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, R.; Lee, Y.G.; Karpurapu, M.; Syed, M.A.; Chung, S.; Deng, J.; Jeong, J.J.; Zhao, G.; Xiao, L.; Sadikot, R.T.; et al. P47phox and reactive oxygen species production modulate expression of microRNA-451 in macrophages. Free Radic. Res. 2014, 49, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Cheng, H.-W.; Qiu, Y.; Dupee, D.; Noonan, M.; Lin, Y.-D.; Fisch, S.; Unno, K.; Sereti, K.-I.; Liao, R. MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ. Res. 2015, 117, 450–459. [Google Scholar] [CrossRef]
- Bulvik, R.; Biton, M.; Berkman, N.; Breuer, R.; Wallach-Dayan, S.B. Forefront: MiR-34a-knockout mice with wild type hematopoietic cells, retain persistent fibrosis following lung injury. Int. J. Mol. Sci. 2020, 21, 2228. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, A.; Ruike, Y.; Terasawa, K.; Tsujimoto, G. MiRNAs and regulation of cell signaling. FEBS J. 2011, 278, 1610–1618. [Google Scholar] [CrossRef]
- Kelleher, Z.T.; Potts, E.N.; Brahmajothi, M.V.; Foster, M.W.; Auten, R.L.; Foster, W.M.; Marshall, H.E. NOS2 regulation of LPS-induced airway inflammation via S-nitrosylation of NF-{kappa}B p65. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L327–L333. [Google Scholar] [CrossRef]
- Asti, C.; Ruggieri, V.; Porzio, S.; Chiusaroli, R.; Melillo, G.; Caselli, G.F. Lipopolysaccharide-induced lung injury in mice. I. Concomitant evaluation of inflammatory cells and haemorrhagic lung damage. Pulm. Pharmacol. Ther. 2000, 13, 61–69. [Google Scholar] [CrossRef]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an active disease biomarker: Recent biochemical and pathological perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2010, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Golubinskaya, V.; Brandt-Eliasson, U.; Gan, L.-M.; Kjerrulf, M.; Nilsson, H. Endothelial function in a mouse model of myeloperoxidase deficiency. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, C.L.; Ford, D.A. MPO (Myeloperoxidase) caused endothelial dysfunction. Arter. Thromb. Vasc. Boil. 2018, 38, 1676–1677. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, C.; Guo, M.; Tao, Y.; Cui, P.; Zhou, Y.; Qin, N.; Zheng, J.; Zhang, J.; Xu, L. MicroRNA-7 deficiency ameliorates the pathologies of acute lung injury through elevating KLF4. Front. Immunol. 2016, 7, 389. [Google Scholar] [CrossRef] [Green Version]
- Almatroodi, S.A.; Almatroudi, A.; Alsahli, M.A.; Aljasir, M.A.; Syed, M.A.; Rahmani, A.H. Epigallocatechin-3-Gallate (EGCG), an active compound of green tea attenuates acute lung injury regulating macrophage polarization and Krüpple-like-factor 4 (KLF4) expression. Molecules 2020, 25, 2853. [Google Scholar] [CrossRef]
- Chen, Q.; Li, L.; Tu, Y.; Zheng, L.L.; Liu, W.; Zuo, X.Y.; He, Y.M.; Zhang, S.Y.; Zhu, W.; Cao, J.P.; et al. MiR-34a regulates apoptosis in liver cells by targeting the KLF4 gene. Cell. Mol. Boil. Lett. 2014, 19, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, M.W.; Cao, Z.; Wara, A.K.; Lebedeva, M.A.; SenBanerjee, S.; Jain, M.K. Kruppel-like Factor 4 is a mediator of proinflammatory signaling in macrophages. J. Boil. Chem. 2005, 280, 38247–38258. [Google Scholar] [CrossRef] [Green Version]
- Tateda, K.; Matsumoto, T.; Miyazaki, S.; Yamaguchi, K. Lipopolysaccharide-induced lethality and cytokine production in aged mice. Infect. Immun. 1996, 64, 769–774. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Hu, S.-J. Effect of microRNA-34a in cell cycle, differentiation, and apoptosis: A review. J. Biochem. Mol. Toxicol. 2011, 26, 79–86. [Google Scholar] [CrossRef]
- Sun, F.; Fu, H.; Liu, Q.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008, 582, 1564–1568. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Barile, G.; Chang, S.; Hays, A.; Pachydaki, S.; Schiff, W.; Sparrow, J. Apoptosis and cell proliferation in proliferative retinal disorders: PCNA, Ki-67, Caspase-3, and PARP expression. Curr. Eye Res. 2005, 30, 395–403. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.J.; Singh, P.; Dohare, R.; Jha, R.; Rahmani, A.H.; Almatroodi, S.A.; Ali, S.; Syed, M.A. Inhibition of miRNA-34a Promotes M2 Macrophage Polarization and Improves LPS-Induced Lung Injury by Targeting Klf4. Genes 2020, 11, 966. https://doi.org/10.3390/genes11090966
Khan MJ, Singh P, Dohare R, Jha R, Rahmani AH, Almatroodi SA, Ali S, Syed MA. Inhibition of miRNA-34a Promotes M2 Macrophage Polarization and Improves LPS-Induced Lung Injury by Targeting Klf4. Genes. 2020; 11(9):966. https://doi.org/10.3390/genes11090966
Chicago/Turabian StyleKhan, Mohd Junaid, Prithvi Singh, Ravins Dohare, Rishabh Jha, Arshad H. Rahmani, Saleh A. Almatroodi, Shakir Ali, and Mansoor Ali Syed. 2020. "Inhibition of miRNA-34a Promotes M2 Macrophage Polarization and Improves LPS-Induced Lung Injury by Targeting Klf4" Genes 11, no. 9: 966. https://doi.org/10.3390/genes11090966
APA StyleKhan, M. J., Singh, P., Dohare, R., Jha, R., Rahmani, A. H., Almatroodi, S. A., Ali, S., & Syed, M. A. (2020). Inhibition of miRNA-34a Promotes M2 Macrophage Polarization and Improves LPS-Induced Lung Injury by Targeting Klf4. Genes, 11(9), 966. https://doi.org/10.3390/genes11090966