The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy

 ,
,  , , ,
, , ,  , , , , , , , ,
, , , , , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Description of the Patients
2.2. Cardiac Magnetic Resonance Imaging
2.3. Molecular Genetic Analysis
2.4. Cloning, Site-Directed Mutagenesis, Cell Culture, and Confocal Microscopy
2.5. Statistical Analysis of Aggregate Formation in Transfected SW-13 Cells
3. Results
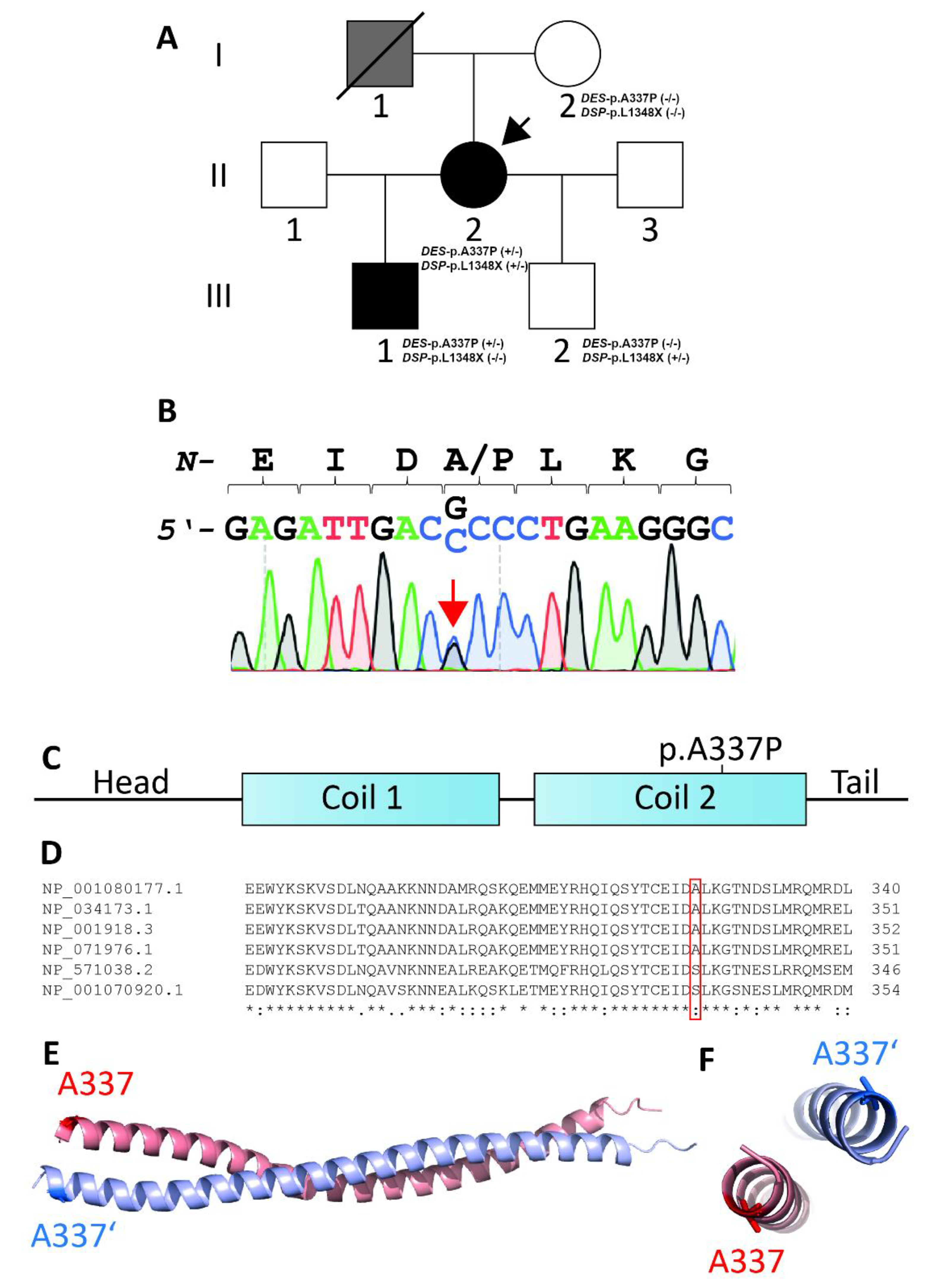
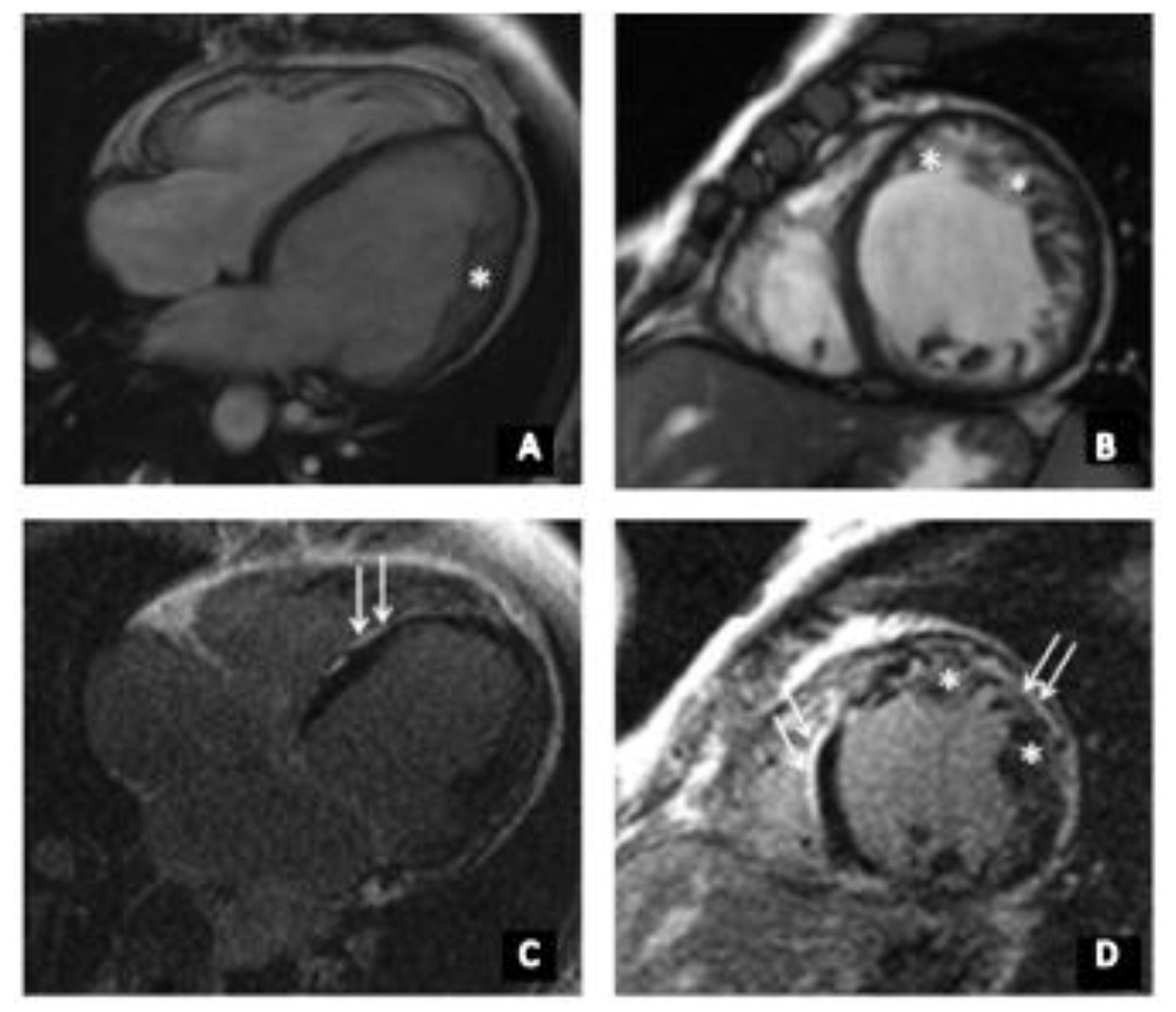
3.1. Clinical Investigations
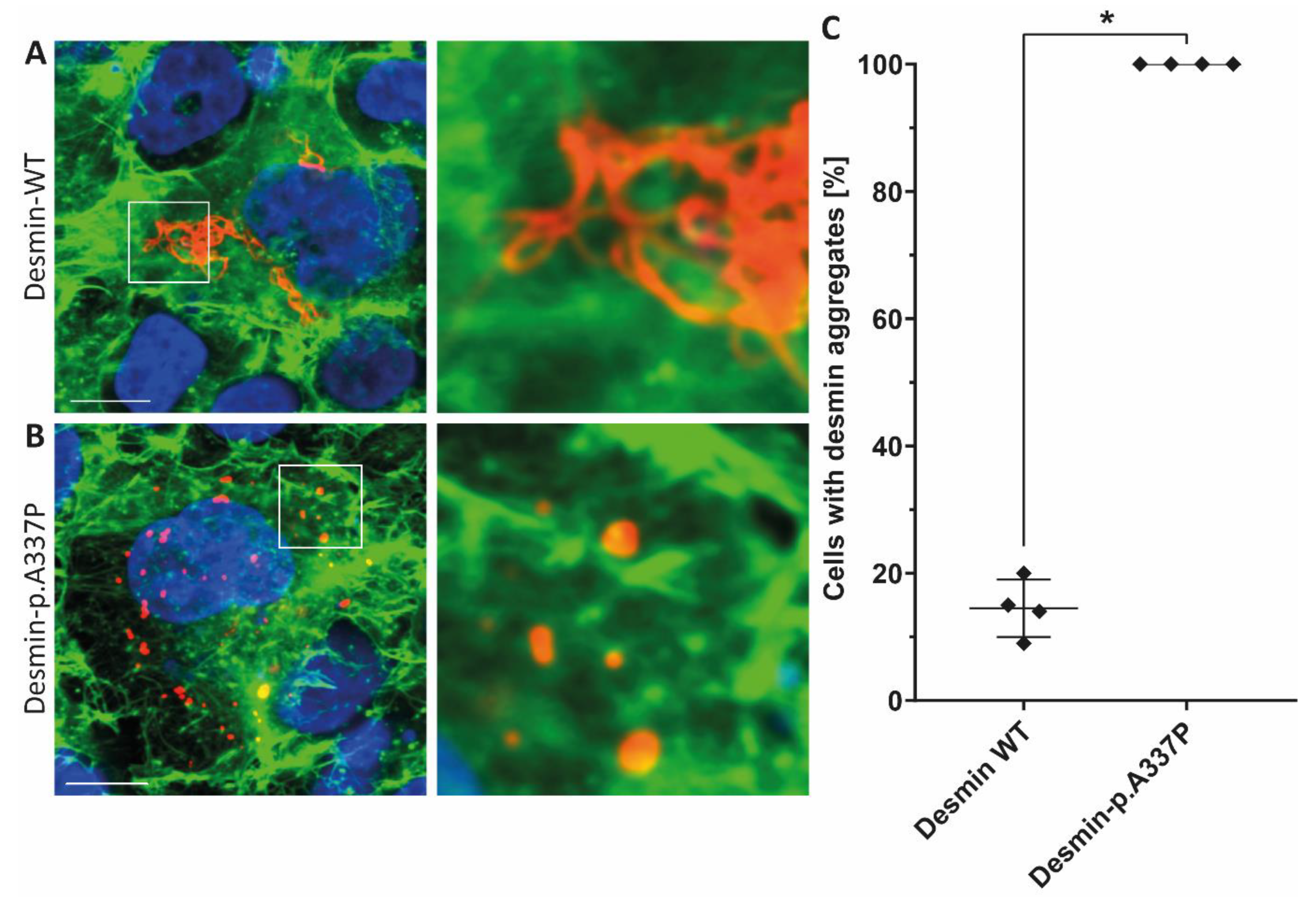
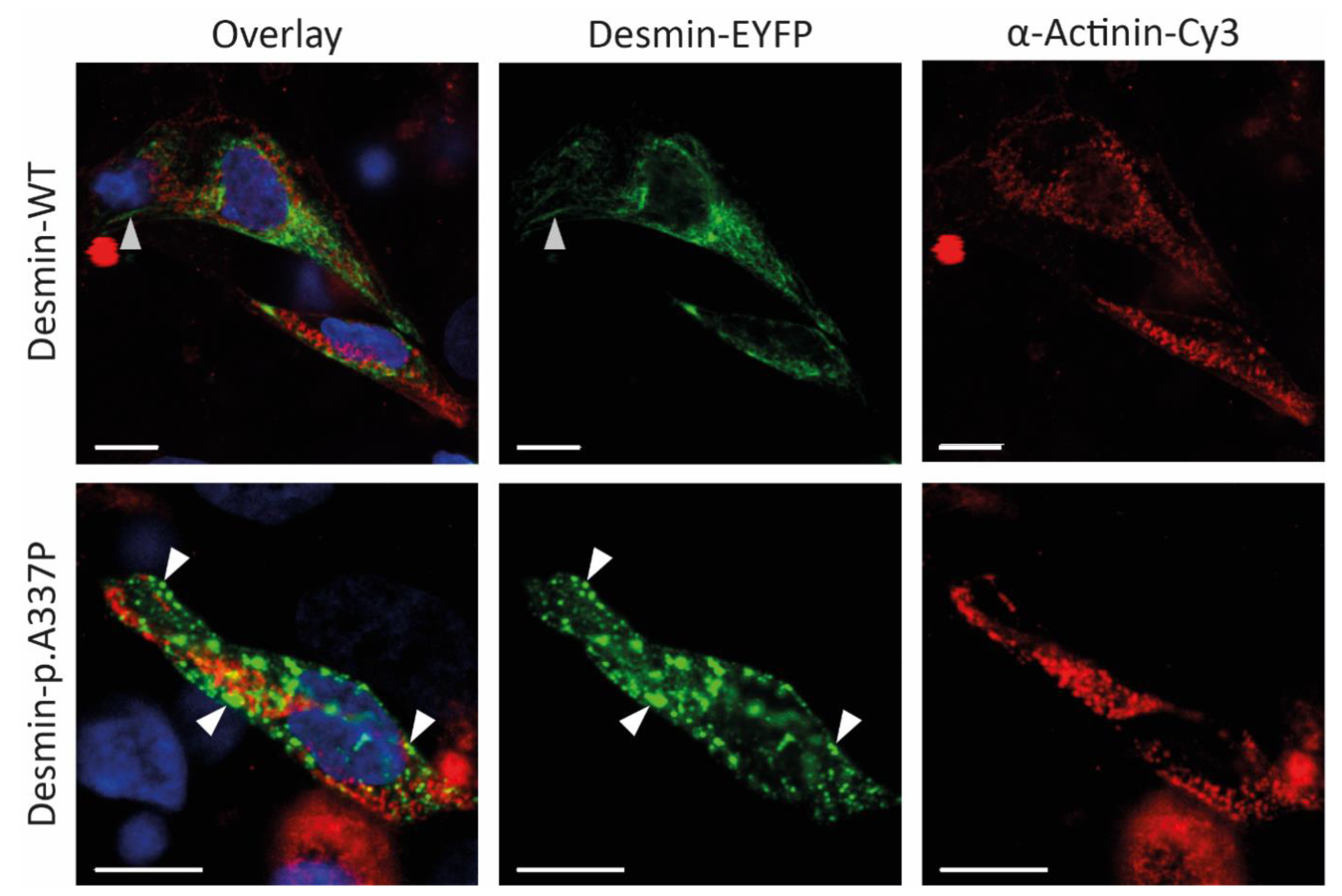
3.2. Genetic and Functional Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Van Waning, J.I.; Moesker, J.; Heijsman, D.; Boersma, E.; Majoor-Krakauer, D. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012993. [Google Scholar] [CrossRef] [PubMed]
- Jenni, R.; Oechslin, E.; Schneider, J.; Attenhofer Jost, C.; Kaufmann, P.A. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart 2001, 86, 666–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Harada, H.; Hayashi, T.; Nishi, H.; Kusaba, K.; Koga, Y.; Koga, Y.; Nonaka, I.; Kimura, A. Phenotypic expression of a novel desmin gene mutation: Hypertrophic cardiomyopathy followed by systemic myopathy. J. Hum. Genet. 2018, 63, 249–254. [Google Scholar] [CrossRef]
- Bermudez-Jimenez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.A.; Alvarez, M.; Lopez-Fernandez, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Cabet, E.; Chevalier, N.R.; Moosmann, J.; Schultheis, D.; Haas, J.; Schowalter, M.; Berwanger, C.; Weyerer, V.; Agaimy, A.; et al. Dual Functional States of R406W-Desmin Assembly Complexes Cause Cardiomyopathy With Severe Intercalated Disc Derangement in Humans and in Knock-In Mice. Circulation 2020, 142, 2155–2171. [Google Scholar] [CrossRef]
- Schirmer, I.; Dieding, M.; Klauke, B.; Brodehl, A.; Gaertner-Rommel, A.; Walhorn, V.; Gummert, J.; Schulz, U.; Paluszkiewicz, L.; Anselmetti, D.; et al. A novel desmin (DES) indel mutation causes severe atypical cardiomyopathy in combination with atrioventricular block and skeletal myopathy. Mol. Genet. Genom. Med. 2018, 6, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubanek, M.; Schimerova, T.; Piherova, L.; Brodehl, A.; Krebsova, A.; Ratnavadivel, S.; Stanasiuk, C.; Hansikova, H.; Zeman, J.; Palecek, T.; et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. J. Clin. Med. 2020, 9, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Miszalski-Jamka, K.; Jefferies, J.L.; Mazur, W.; Glowacki, J.; Hu, J.; Lazar, M.; Gibbs, R.A.; Liczko, J.; Klys, J.; Venner, E.; et al. Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients With Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 2017, 10, e001763. [Google Scholar] [CrossRef] [Green Version]
- Tamiya, R.; Saito, Y.; Fukamachi, D.; Nagashima, K.; Aizawa, Y.; Ohkubo, K.; Hatta, T.; Sezai, A.; Tanaka, M.; Ishikawa, T.; et al. Desmin-related myopathy characterized by non-compaction cardiomyopathy, cardiac conduction defect, and coronary artery dissection. ESC Heart Fail. 2020, 7, 1338–1343. [Google Scholar] [CrossRef] [Green Version]
- Bar, H.; Mucke, N.; Kostareva, A.; Sjoberg, G.; Aebi, U.; Herrmann, H. Severe muscle disease-causing desmin mutations interfere with in vitro filament assembly at distinct stages. Proc. Natl. Acad. Sci. USA 2005, 102, 15099–15104. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Saric, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [Green Version]
- Heffler, J.; Shah, P.P.; Robison, P.; Phyo, S.; Veliz, K.; Uchida, K.; Bogush, A.; Rhoades, J.; Jain, R.; Prosser, B.L. A Balance Between Intermediate Filaments and Microtubules Maintains Nuclear Architecture in the Cardiomyocyte. Circ. Res. 2020, 126, e10–e26. [Google Scholar] [CrossRef]
- Kedia, N.; Arhzaouy, K.; Pittman, S.K.; Sun, Y.; Batchelor, M.; Weihl, C.C.; Bieschke, J. Desmin forms toxic, seeding-competent amyloid aggregates that persist in muscle fibers. Proc. Natl. Acad. Sci. USA 2019, 116, 16835–16840. [Google Scholar] [CrossRef] [Green Version]
- Goldfarb, L.G.; Park, K.Y.; Cervenakova, L.; Gorokhova, S.; Lee, H.S.; Vasconcelos, O.; Nagle, J.W.; Semino-Mora, C.; Sivakumar, K.; Dalakas, M.C. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 1998, 19, 402–403. [Google Scholar] [CrossRef]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left Ventricular Non-Compaction. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Dieding, M.; Biere, N.; Unger, A.; Klauke, B.; Walhorn, V.; Gummert, J.; Schulz, U.; Linke, W.A.; Gerull, B.; et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J. Mol. Cell. Cardiol. 2016, 91, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gartner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Chernyatina, A.A.; Strelkov, S.V. Stabilization of vimentin coil2 fragment via an engineered disulfide. J. Struct. Biol. 2012, 177, 46–53. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 20. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R. WS-SNPs&GO: A web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genom. 2013, 14, S6. [Google Scholar]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarria, A.J.; Lieber, J.G.; Nordeen, S.K.; Evans, R.M. The presence or absence of a vimentin-type intermediate filament network affects the shape of the nucleus in human SW-13 cells. J. Cell Sci. 1994, 107, 1593–1607. [Google Scholar] [PubMed]
- Li, D.; Tapscoft, T.; Gonzalez, O.; Burch, P.E.; Quinones, M.A.; Zoghbi, W.A.; Hill, R.; Bachinski, L.L.; Mann, D.L.; Roberts, R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999, 100, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Stollberger, C.; Blazek, G.; Wegner, C.; Finsterer, J. Heart failure, atrial fibrillation and neuromuscular disorders influence mortality in left ventricular hypertrabeculation/noncompaction. Cardiology 2011, 119, 176–182. [Google Scholar] [CrossRef]
- Stollberger, C.; Blazek, G.; Wegner, C.; Winkler-Dworak, M.; Finsterer, J. Neuromuscular and cardiac comorbidity determines survival in 140 patients with left ventricular hypertrabeculation/noncompaction. Int. J. Cardiol. 2011, 150, 71–74. [Google Scholar] [CrossRef]
- van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.E.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef]
- Alcalai, R.; Metzger, S.; Rosenheck, S.; Meiner, V.; Chajek-Shaul, T. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J. Am. Coll. Cardiol. 2003, 42, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ayala, J.M.; Gomez-Milanes, I.; Sanchez Munoz, J.J.; Ruiz-Espejo, F.; Ortiz, M.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Oliva-Sandoval, M.J.; Monserrat, L.; Valdes, M.; et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Characterizing a phenotype. Europace 2014, 16, 1838–1846. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Genomic Coordinates | Transcript; DNA Change; Protein Change | dbSNP ID | gnomAD MAF (v. 2.1.1) | ACMG Interpretation | Familial Aggregation with LVNC | The Presence of the Variants in A Family Member | |||
|---|---|---|---|---|---|---|---|---|---|---|
| I-2 | II-2 | III-1 | III-2 | |||||||
| DSP | hg19:chr6:7580464_AC/A | NM_004415.3; c.4042delC; p.Leu1348Ter | - | - | likely pathogenic(PVS1, PM2, BS4) | no | 0/0 | 1/0 | 0/0 | 1/0 |
| DES | hg19:chr2:220285661_G/C | NM_001927.3; c.1009G>C; p.Ala337Pro | rs59962885 | - | likely pathogenic(PS3, PM2. PP1, PP3, PP5) | yes | 0/0 | 1/0 | 1/0 | 0/0 |
| PKP2 | hg19:chr12:32994058_A/C | NM_004572.3; c.1592T>G; p.Ile531Ser | rs147240502 | 0.004746 | likely benign (PM1, PP1, BS1, BP6) | yes | N/A | 1/0 | 1/0 | n/a |
| MYH7 | hg19:chr20:33581180_G/A | NM_020884.4; c.2477G>A; p.Arg826His | rs187028260 | 0.002979 | Benign (PM1, PP3, BS1, BS2) | no | N/A | 1/0 | 0/0 | n/a |
| TTN | hg19:chr2:179395554_GC/AA | NM_001256850.1; c.100864_100865delGCinsTT; p.Ala33622Phe | rs794729250 | - | likely benign (PM2, PP3, BS4, BP1, BP6) | no | N/A | 1/0 | 0/0 | n/a |
| DES | hg19:chr2:220284876_C/T | NM_001927.3; c.638C>T; p.Ala213Val | rs41272699 | 0.009280 | likely benign (PP1, PP3, BS1, BP6) | yes | N/A | 1/0 | 1/0 | n/a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gärtner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. https://doi.org/10.3390/genes12010121
Kulikova O, Brodehl A, Kiseleva A, Myasnikov R, Meshkov A, Stanasiuk C, Gärtner A, Divashuk M, Sotnikova E, Koretskiy S, et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes. 2021; 12(1):121. https://doi.org/10.3390/genes12010121
Chicago/Turabian StyleKulikova, Olga, Andreas Brodehl, Anna Kiseleva, Roman Myasnikov, Alexey Meshkov, Caroline Stanasiuk, Anna Gärtner, Mikhail Divashuk, Evgeniia Sotnikova, Sergey Koretskiy, and et al. 2021. "The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy" Genes 12, no. 1: 121. https://doi.org/10.3390/genes12010121
APA StyleKulikova, O., Brodehl, A., Kiseleva, A., Myasnikov, R., Meshkov, A., Stanasiuk, C., Gärtner, A., Divashuk, M., Sotnikova, E., Koretskiy, S., Kharlap, M., Kozlova, V., Mershina, E., Pilus, P., Sinitsyn, V., Milting, H., Boytsov, S., & Drapkina, O. (2021). The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes, 12(1), 121. https://doi.org/10.3390/genes12010121