DIANA-mAP: Analyzing miRNA from Raw NGS Data to Quantification


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Utilized Resources
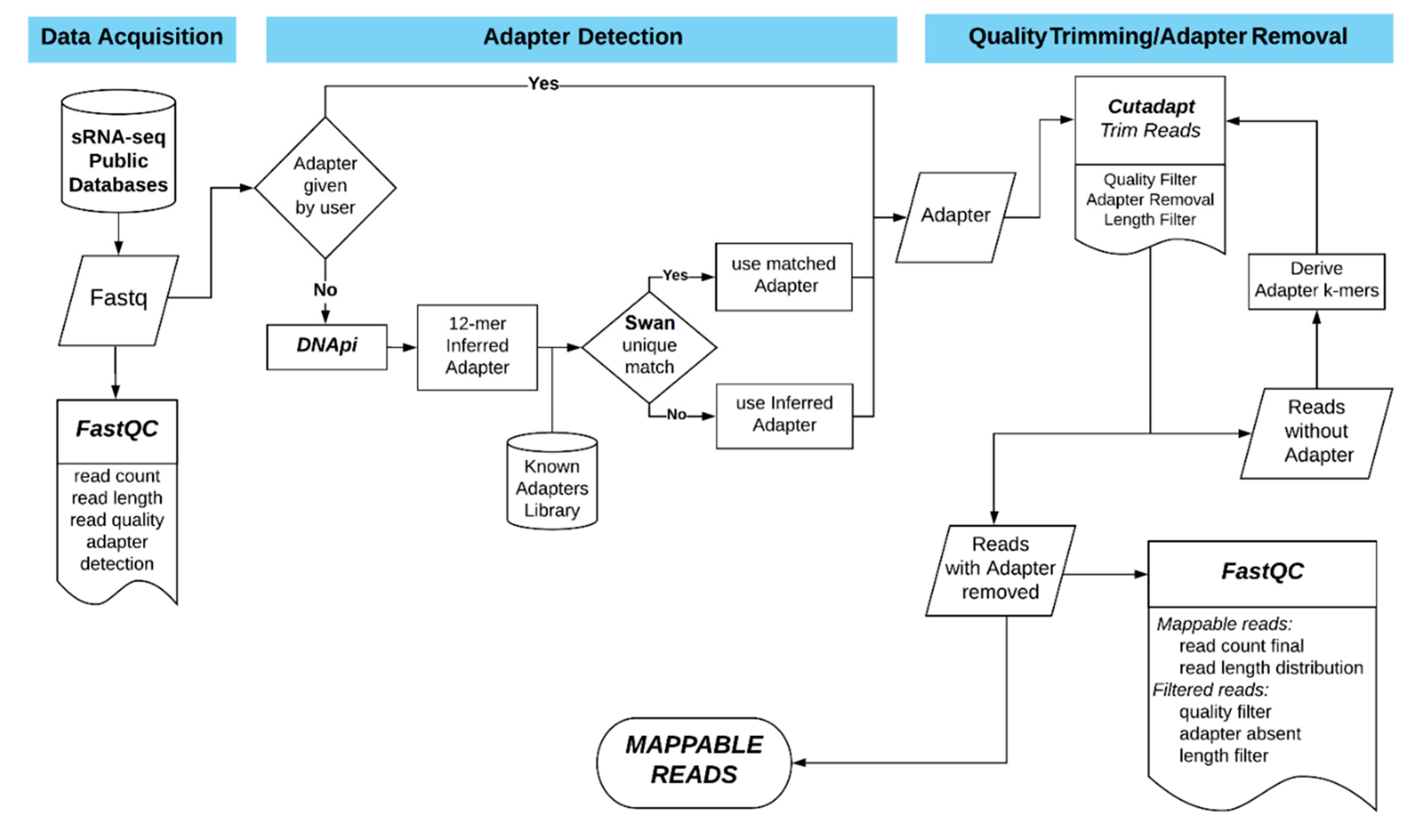
2.2. The DIANA-mAP Analysis Pipeline
2.2.1. Data Acquisition and Preprocessing
2.2.2. Alignment and Quantification
2.2.3. Differential Expression
2.2.4. Results
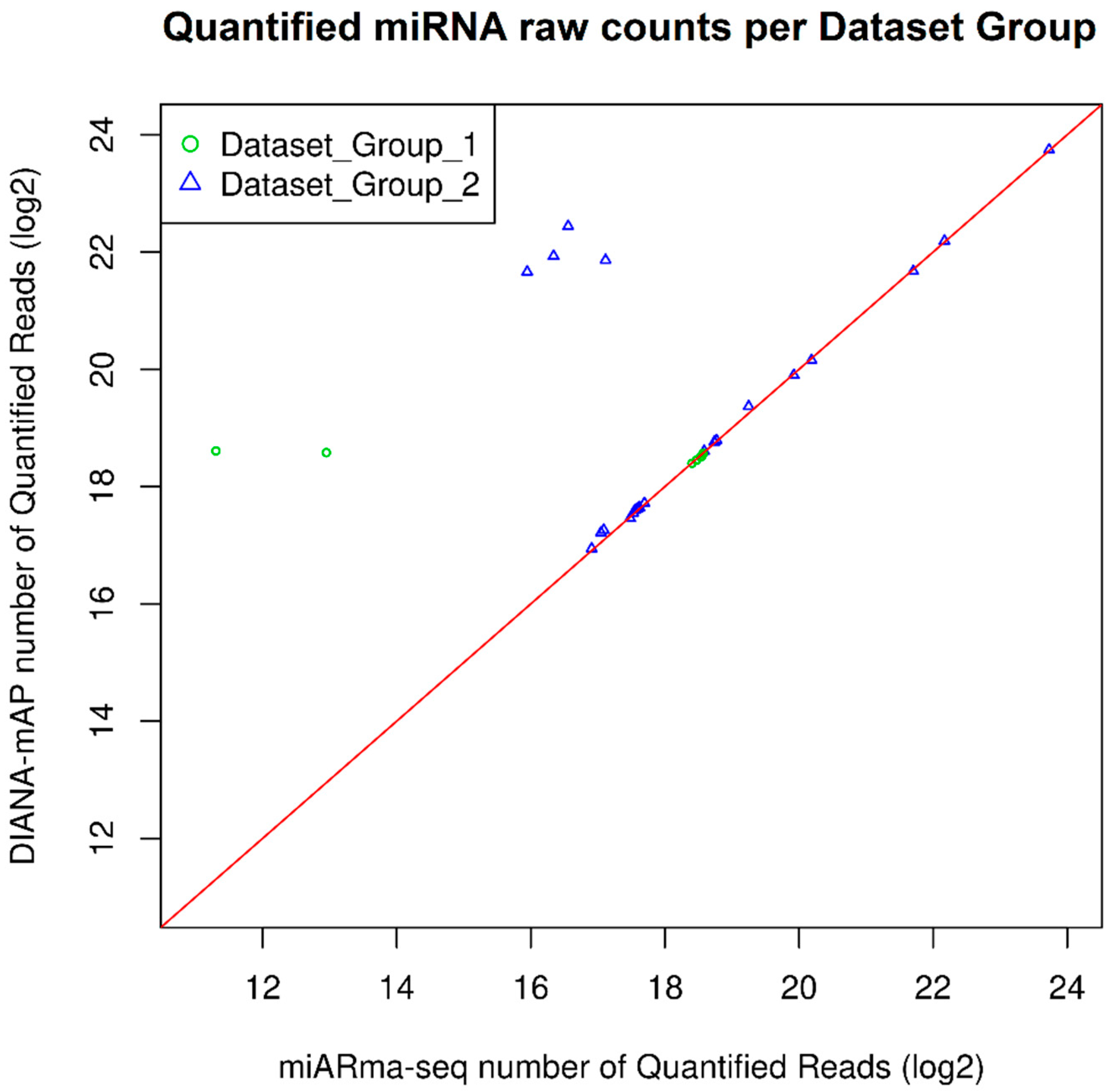
3. Evaluation
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Vlachos, I.; Hatzigeorgiou, A.G. Online resources for miRNA analysis. Clin. Biochem. 2013, 46, 879–900. [Google Scholar] [CrossRef]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 7385. [Google Scholar] [CrossRef]
- Zhang, C. Novel functions for small RNA molecules. Curr. Opin. Mol. Ther. 2009, 11, 641–651. [Google Scholar]
- Wu, X.; Kim, T.-K.; Baxter, D.; Scherler, K.; Gordon, A.; Fong, O.; Etheridge, A.; Galas, D.J.; Wang, K. sRNAnalyzer—A flexible and customizable small RNA sequencing data analysis pipeline. Nucleic Acids Res. 2017, 45, 12140–12151. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Gordon, W.; Du, S.; Zhang, C.; He, W.; Xi, L.; Mathur, S.; Agostino, M.; Paradis, T.; Von Schack, D.; et al. QuickMIRSeq: A pipeline for quick and accurate quantification of both known miRNAs and isomiRs by jointly processing multiple samples from microRNA sequencing. BMC Bioinform. 2017, 18, 180. [Google Scholar] [CrossRef] [Green Version]
- Desvignes, T.; Batzel, P.; Sydes, J.; Eames, B.F.; Postlethwait, J.H. miRNA analysis with Prost! reveals evolutionary conservation of organ-enriched expression and post-transcriptional modifications in three-spined stickleback and zebrafish. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Pla, A.; Rayner, S. Jasmine: A Java pipeline for isomiR characterization in miRNA-Seq data. Bioinformatics 2020, 36, 1933–1936. [Google Scholar] [CrossRef] [Green Version]
- Rueda, A.; Barturen, G.; Lebrón, R.; Gómez-Martín, C.; Alganza, Á.; Oliver, J.L.; Hackenberg, M. sRNAtoolbox: An integrated collection of small RNA research tools. Nucleic Acids Res. 2015, 43, W467–W473. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, Q.; Wang, X.; Zheng, J.; Wang, T.; You, M.; Sun, Z.S.; Shi, Q. mirTools 2.0 for non-coding RNA discovery, profiling, and functional annotation based on high-throughput sequencing. RNA Biol. 2013, 10, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Evans, J.M.; Bhagwate, A.V.; Middha, S.; Bockol, M.; Yan, H.; Kocher, J.-P.A. CAP-miRSeq: A comprehensive analysis pipeline for microRNA sequencing data. BMC Genom. 2014, 15, 423. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Baras, A.S.; Halushka, M.K. miRge 2.0 for comprehensive analysis of microRNA sequencing data. BMC Bioinform. 2018, 19, 275. [Google Scholar] [CrossRef]
- Andrés-León, E.; Núñez-Torres, R.; Rojas, A.M. miARma-Seq: A comprehensive tool for miRNA, mRNA and circRNA analysis. Sci. Rep. 2016, 6, 25749. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.P.; Van Dongen, S.; Abreu-Goodger, C.; Bartonicek, N.; Enright, A.J. Kraken: A set of tools for quality control and analysis of high-throughput sequence data. Methods 2013, 63, 41–49. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; On Behalf of the International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011, 39 (Suppl. 1), D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Springer: New York, NY, USA, 2016; pp. 93–110. [Google Scholar]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39 (Suppl. 1), D152–D157. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Tsuji, J.; Weng, Z. DNApi: A De Novo Adapter Prediction Algorithm for Small RNA Sequencing Data. PLoS ONE 2016, 11, e0164228. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Adapter Trimming: Why Are Adapter Sequences Trimmed from only the 3’ Ends of Reads? Available online: https://emea.support.illumina.com/bulletins/2016/04/adapter-trimming-why-are-adapter-sequences-trimmed-from-only-the--ends-of-reads.html (accessed on 6 November 2020).
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Farazi, T.A.; Horlings, H.M.; Hoeve, J.J.T.; Mihailovic, A.; Halfwerk, H.; Morozov, P.; Brown, M.; Hafner, M.; Reyal, F.; Van Kouwenhove, M.; et al. MicroRNA Sequence and Expression Analysis in Breast Tumors by Deep Sequencing. Cancer Res. 2011, 71, 4443–4453. [Google Scholar] [CrossRef] [Green Version]
- Camps, C.; Saini, H.K.; Mole, D.R.; Choudhry, H.; Reczko, M.; Guerra-Assunção, J.A.; Tian, Y.-M.; Buffa, F.M.; Harris, A.L.; Hatzigeorgiou, A.G.; et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol. Cancer 2014, 13, 28. [Google Scholar] [CrossRef] [Green Version]
- Jima, D.D.; Zhang, J.; Jacobs, C.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.L.; Au, W.Y.; Srivastava, G.; Czader, M.B.; Rizzieri, D.A.; et al. Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood 2010, 116, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, I.; Vergoulis, T.; Paraskevopoulou, M.D.; Lykokanellos, F.; Georgakilas, G.; Georgiou, P.; Chatzopoulos, S.; Karagkouni, D.; Christodoulou, F.; Dalamagas, T.; et al. DIANA-mirExTra v2.0: Uncovering microRNAs and transcription factors with crucial roles in NGS expression data. Nucleic Acids Res. 2016, 44, W128–W134. [Google Scholar] [CrossRef]
- Handzlik, J.E.; Tastsoglou, S.; Vlachos, I.; Hatzigeorgiou, A. Manatee: Detection and quantification of small non-coding RNAs from next-generation sequencing data. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset Group 1 | |||||||
|---|---|---|---|---|---|---|---|
| Dataset Accession No. | Number of Reads | Mapped Reads | Quantified Reads | ||||
| miARma-Seq | DIANA-mAP | Difference (% of Total Reads) | miARma-Seq | DIANA-mAP | Difference (% of Total Reads) | ||
| SRR873382 | 500000 | 410190 (82.04%) | 395314 (79.06%) | 2.98 | 384692 (76.94%) | 377084 (75.42%) | 1.52 |
| SRR873383 | 500000 | 404071 (80.81%) | 388416 (77.68%) | 3.13 | 379041 (75.81%) | 371648 (74.33%) | 1.48 |
| SRR873384 | 500000 | 376218 (75.24%) | 368605 (73.72%) | 1.52 | 363215 (72.64%) | 358021 (71.60%) | 1.04 |
| SRR873385 | 500000 | 365339 (73.07%) | 359338 (71.87%) | 1.2 | 346597 (69.32%) | 345055 (69.01%) | 0.31 |
| SRR873386 | 500000 | 10467 (2.09%) | 406743 (81.35%) | 79.26 | 7915 (1.58%) | 391566 (78.31%) | 76.73 |
| SRR873387 | 500000 | 416391 (83.28%) | 406197 (81.24%) | 2.04 | 389770 (77.95%) | 389028 (77.81%) | 0.15 |
| SRR873388 | 500000 | 408720 (81.74%) | 400364 (80.07%) | 1.67 | 389295 (77.86%) | 386969 (77.39%) | 0.47 |
| SRR873389 | 500000 | 3182 (0.64%) | 415122 (83.02%) | 82.39 | 2529 (0.51%) | 399397 (79.88%) | 79.37 |
| Quantified miRNA Reads | % of Total Reads | |
|---|---|---|
| Simulated Reads | 308868 | 39.7 |
| DIANA-mAP | 300842 | 38.7 |
| miARma-Seq | 260821 | 33.5 |
| Pearson Correlation Coefficient | Pearson p-Value | |
|---|---|---|
| DIANA-mAP vs. Simulated Reads | 0.9602398 | <2.2 × 10−16 |
| miARma-Seq vs. Simulated Reads | 0.9376623 | <2.2 × 10−16 |
| DIANA-mAP vs. miARma-Seq | 0.9231243 | <2.2 × 10−16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexiou, A.; Zisis, D.; Kavakiotis, I.; Miliotis, M.; Koussounadis, A.; Karagkouni, D.; Hatzigeorgiou, A.G. DIANA-mAP: Analyzing miRNA from Raw NGS Data to Quantification. Genes 2021, 12, 46. https://doi.org/10.3390/genes12010046
Alexiou A, Zisis D, Kavakiotis I, Miliotis M, Koussounadis A, Karagkouni D, Hatzigeorgiou AG. DIANA-mAP: Analyzing miRNA from Raw NGS Data to Quantification. Genes. 2021; 12(1):46. https://doi.org/10.3390/genes12010046
Chicago/Turabian StyleAlexiou, Athanasios, Dimitrios Zisis, Ioannis Kavakiotis, Marios Miliotis, Antonis Koussounadis, Dimitra Karagkouni, and Artemis G. Hatzigeorgiou. 2021. "DIANA-mAP: Analyzing miRNA from Raw NGS Data to Quantification" Genes 12, no. 1: 46. https://doi.org/10.3390/genes12010046
APA StyleAlexiou, A., Zisis, D., Kavakiotis, I., Miliotis, M., Koussounadis, A., Karagkouni, D., & Hatzigeorgiou, A. G. (2021). DIANA-mAP: Analyzing miRNA from Raw NGS Data to Quantification. Genes, 12(1), 46. https://doi.org/10.3390/genes12010046