The Developmental Transcriptome of Bagworm, Metisa plana (Lepidoptera: Psychidae) and Insights into Chitin Biosynthesis Genes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Library Construction and Sequencing
2.3. Transcriptome Assembly
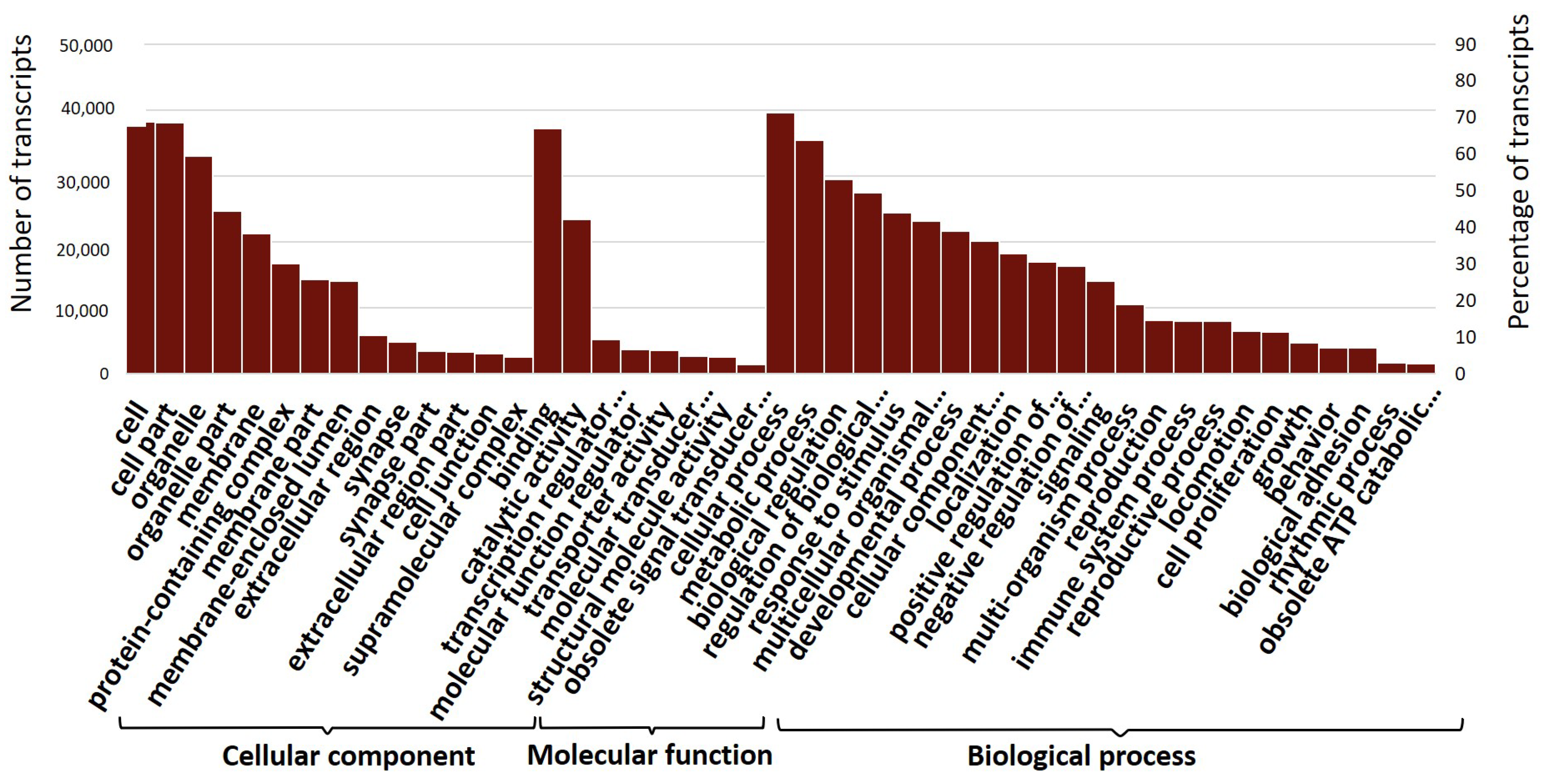
2.4. Transcriptome Annotation and Gene Ontology
2.5. Analysis of Differentially Expressed Transcripts (DETs)
2.6. SSR and SNPs Mining
3. Results
3.1. Sequencing and de novo Assembly
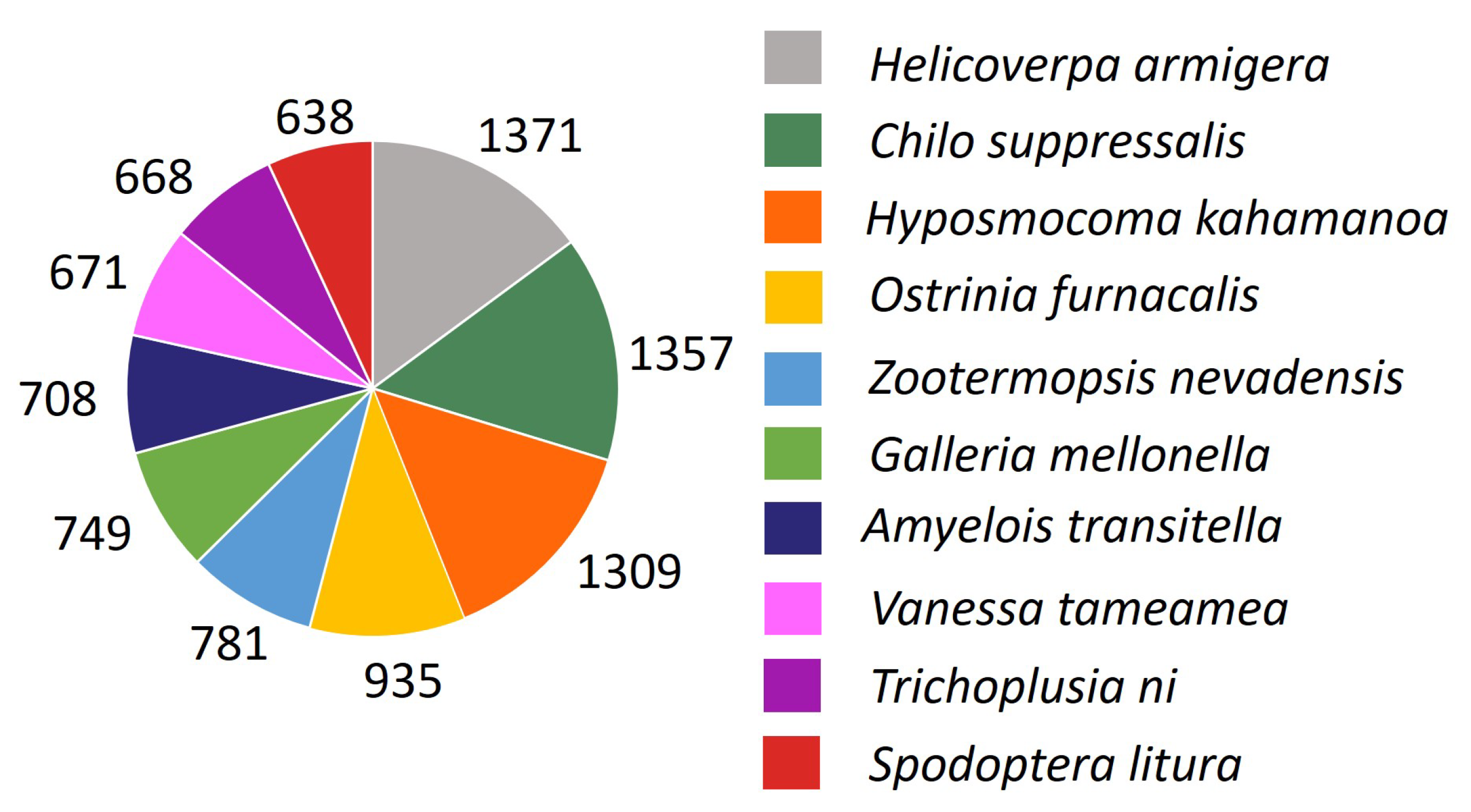
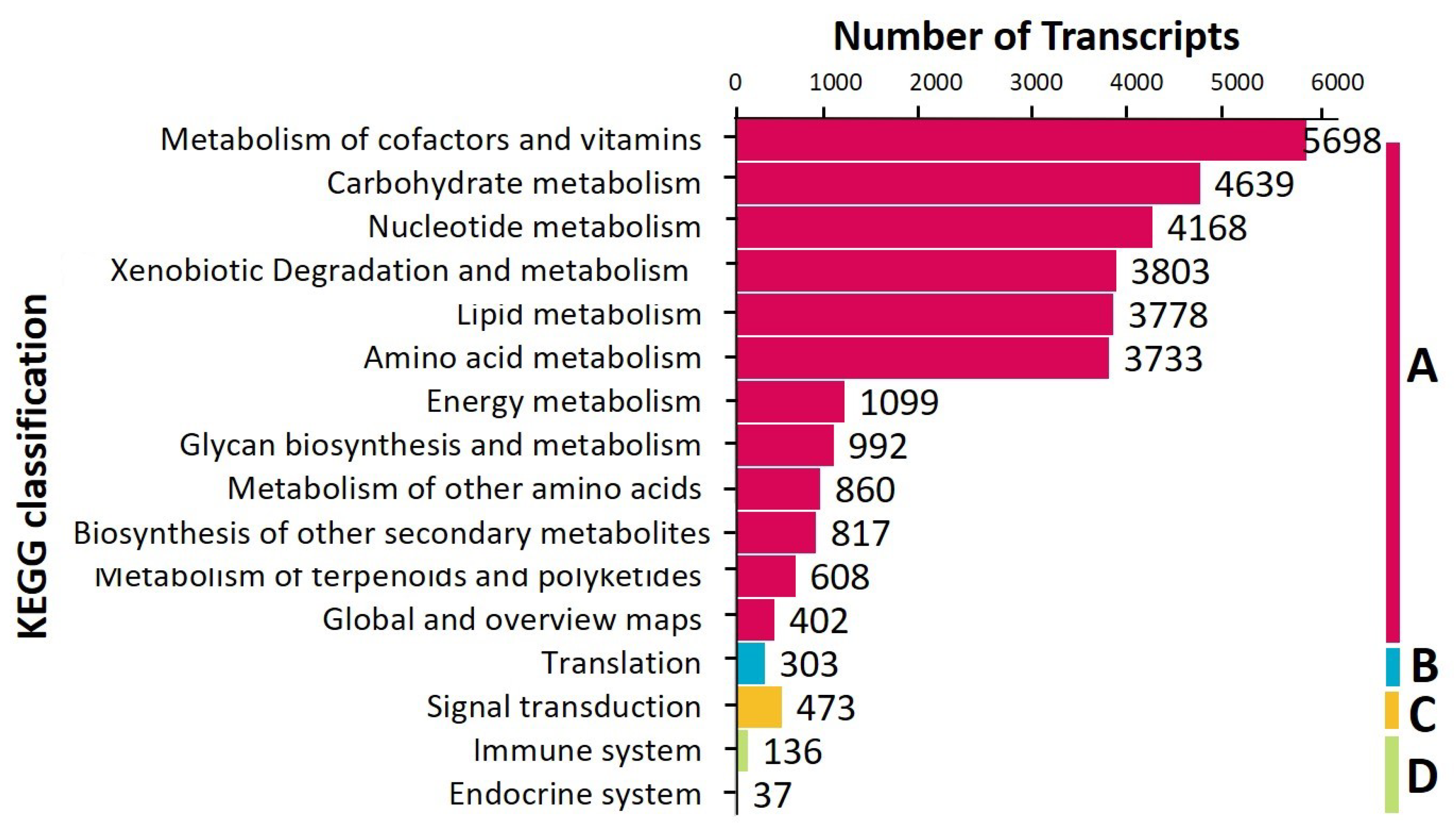
3.2. Annotation and KEGG Classification
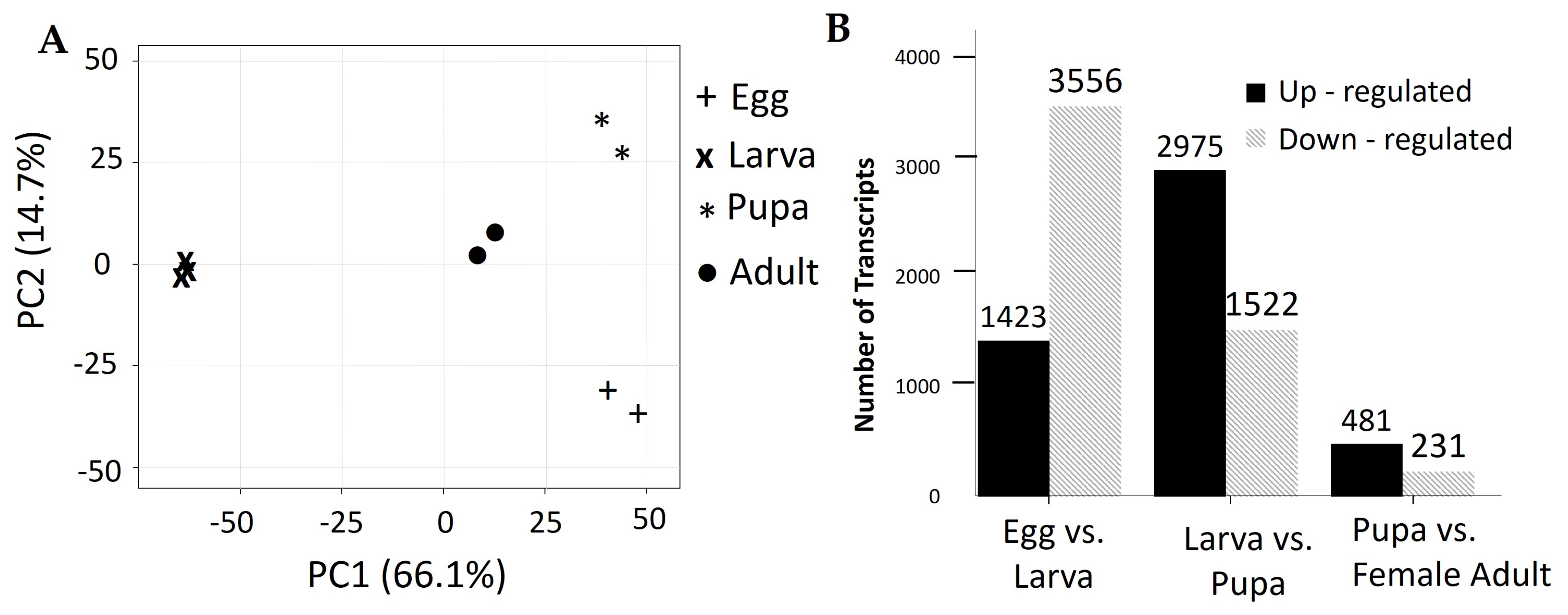
3.3. Analysis of Differentially Expressed Transcripts
3.4. Expression Profile and Enrichment Analysis
3.5. Identification and Expression Analysis of Transcripts Encoding Chitin Biosynthesis
3.6. SSR and SNPs Mining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oil Palm Planted Area 2018. Available online: http://bepi.mpob.gov.my/images/area/2018/Area_summary.pdf (accessed on 7 January 2020).
- Palm Oil Market Share Expected to Cross USD 92.84 Billion by 2021. Available online: https://www.globenewswire.com/news-release/2017/11/21/1197983/0/en/Palm-Oil-Market-Share-Expected-to-Cross-USD-92-84-Billion-by-2021-Zion-Market-Research.html (accessed on 7 January 2020).
- Kok, C.C.; Eng, O.K.; Razak, A.R.; Arshad, A.M. Microstructure and life cycle of Metisa plana Walker (Lepidoptera: Psychidae). J. Sustain. Sci. Manag. 2011, 6, 51–59. [Google Scholar]
- Dziadik-Turner, C.; Koga, D.; Mai, M.S.; Kramer, K. Purification and characterization of two β-N-Acetylhexosaminidases from the Tobacco Hornworm, Manduca sexta (L.) (Lepidoptera:Sphingidae). Arch. Biochem. Biophys. 1981, 212, 546–560. [Google Scholar] [CrossRef]
- Ou, J.; Deng, H.M.; Zheng, S.C.; Huang, L.H.; Feng, Q.L.; Liu, L. Transcriptomic analysis of developmental features of Bombyx mori wing disc during metamorphosis. BMC Genom. 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussian, B.; Schwarz, H.; Bartoszewski, S.; Nüsslein-Volhard, C. Involvement of chitin in exoskeleton morphogenesis in Drosophila melanogaster. J. Morphol. 2005, 264, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Maas, W.; Van Hes, R.; Grosscurt, A.C.; Deul, D.H. Benzoylphenyl urea insecticides. In Chemie der Pfl Anzenschutzund Schadlingsbekampfungsmittel; Wegler, R., Ed.; Springer: Berlin/Heidelberg, Germany, 1981; pp. 423–470. [Google Scholar]
- Campbell, B.; Baldwin, R.; Koehler, P. Locomotion inhibition of Cimex lectularius L. following topical, sublethal dose application of the chitin synthesis inhibitor Lufenuron. Insects 2017, 8, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belinato, T.; Martins, A.J.; Lima, J.B.P.; Valle, D. Effect of Triflumuron, a chitin synthesis inhibitor, on Aedes aegypti, Aedes albopictus and Culex quinquefasciatus under laboratory conditions. Parasites Vectors 2013, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.; Aljabr, A.M.; Al-Ayedh, H. Development-disrupting chitin synthesis inhibitor, Novaluron, reprogramming the chitin degradation mechanism of Red Palm Weevils. Molecules 2019, 24, 4304. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.A.; Lacerda, M.C.; Zanuncio, T.V.; Ramalho, F.D.S.; Polanczyk, R.A.; Serrão, J.E.; Zanuncio, J.C. Effect of the insect growth regulator diflubenzuron on the predator Podisus nigrispinus (Heteroptera: Pentatomidae). Ecotoxicology 2012, 21, 96–103. [Google Scholar] [CrossRef]
- Mansur, J.F.; Figueira-Mansur, J.; Santos, A.S.; Santos-Junior, H.; Ramos, I.B.; de Medeiros, M.N.; Machado, E.A.; Kaiser, C.R.; Muthukrishnan, S.; Masuda, H.; et al. The effect of Lufenuron, a chitin synthesis inhibitor, on oogenesis of Rhodnius prolixus. Pestic. Biochem. Physiol. 2010, 98, 59–67. [Google Scholar] [CrossRef]
- Halim, M.; Muhaimin, A.M.D.; Syarifah Zulaikha, S.A.; Nor Atikah, A.R.; Masri, M.M.M.; Yaakop, S. Evaluation of infestation in parasitoids on Metisa plana Walker (Lepidoptera: Psychidae) in three oil palm plantations in Peninsular Malaysia. Serangga 2017, 22, 135–149. [Google Scholar]
- Salim, H.; Hamid, N.H. Evaluation of several chemical control approaches against bagworm, Metisa plana Walker (Lepidopterea: Psychidae) in Felda oil palm plantations. Plant 2012, 88, 785–799. [Google Scholar]
- Kamarudin, N.; Ali, S.R.A.; Masri, M.M.M.; Ahmad, M.N.; Manan, C.A.H.C.; Kamarudin, N. Controlling Metisa plana Walker (Lepidoptera: Psychidae) outbreak Using Bacillus thuringiensis at an oil palm plantation in Slim River, Perak, Malaysia. J. Oil Palm Res. 2017, 29, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.; Kamarudin, N.; Ahmad, S.N.; Arshad, O.; Mohd Masri, M.M.; Moslim, R.; Kushairi, A. Efficacy of pheromone trapping and aerial spraying of Bacillus Thuringiensis (Bt) for controlling bagworm, Metisa plana (Lepidoptera: Psychidae). J. Oil Palm Res. 2017, 29, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Halim, M.; Aman-Zuki, A.; Syed-Ahmad, S.Z.; Mohammad-Din, A.M.; Abdul-Rahim, A.; Mohd-Masri, M.M.; Md-Zain, B.M.; Yaakop, S. Exploring the abundance and DNA barcode information of eight parasitoid wasps species (Hymenoptera), the natural enemies of the important pest of oil palm, bagworm, Metisa plana (Lepidoptera: Psychidae) toward the biocontrol approach and it’s application. J. Asia. Pac. Entomol. 2018, 21, 1359–1365. [Google Scholar] [CrossRef]
- Kamarudin, N.; Ahmad, S.N.; Arshad, O.; Wahid, M.B. Pheromone mass trapping of bagworm moths, Metisa plana Walker (Lepidoptera: Psychidae), for its control in mature oil palms in Perak, Malaysia. J. Asia. Pac. Entomol. 2010, 13, 101–106. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA Using TRIzol (TRI Reagent); Cold Spring Harbor Lab Press: Cold Spring, NY, USA, 2010. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. 2010. [Google Scholar]
- Cai, L.; Li, J.; Yu, L.; Wei, Y.; Miao, Z.; Chen, M.; Huang, R. Marine genomics de novo transcriptome assembly of the new marine fish model of Goby, Mugilogobius chulae. Mar. Genom. 2018, 40, 18–20. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Ido, A.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2013, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.; Giang, T.; Van Nguyen, L.; Quang, H.; Hai, T. De novo assembly and transcriptome characterization of major growth-related genes in various tissues of Penaeus monodon. Aquaculture 2016, 464, 545–553. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 46. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Philip, D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Macmanes, M.D.; et al. De novo transcript sequence recostruction from RNA-Seq: Reference generation and analysis with trinity. Nat. Protoc. 2013, 8, 1–43. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 1999, 27, 49–54. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, F.; Rieske, L.K.; Sun, L.L.; Sun, S.H. Transcriptome sequencing for identification of diapause-associated genes in fall webworm, Hyphantria cunea Drury. Gene 2018, 668, 229–236. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Sacan, A. Weighted set enrichment of gene expression data. BMC Syst. Biol. 2013, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aickin, M.; Gensler, H. Adjusting for multiple testing when reporting research results: The Bonferroni vs Holm methods. Am. J. Public Health 1996, 86, 726–728. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ping, J.; Chen, H.; Jiao, L.; Zheng, S.; Han, Z.G.; Hao, P.; Huang, J. A comparative analysis of liver transcriptome suggests divergent liver function among human, mouse and rat. Genomics 2010, 96, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, P.; Luo, Z. GMATo: A novel tool for the identification and analysis of microsatellites in large genomes. Bioinformation 2013, 9, 541–544. [Google Scholar] [CrossRef] [Green Version]
- The GATK Best Practices for Variant Calling on RNAseq. Available online: https://rna-seqblog.com/the-gatk-best-practices-for-variant-calling-on-rnaseq/ (accessed on 19 October 2020).
- Van der Auwera, G.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Merzendorfer, H.; Zimoch, L. Chitin metabolism in insects: Structure, function and regulation of chitin synthases and chitinases. J. Exp. Biol. 2003, 206, 4393–4412. [Google Scholar] [CrossRef] [Green Version]
- Kramer, K.J.; Koga, D. Insect chitin: Physical state, synthesis, degradation and metabolic regulation. Insect Biochem. 1986, 16, 851–877. [Google Scholar] [CrossRef]
- Cohen, E. chitin synthesis and inhibition: A revisit. Pest Manag. Sci. 2000, 57, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yang, P.C.; Li, J.; Yang, F.; Zhang, A.B. Transcriptome characterization of Dendrolimus punctatus and expression profiles at different developmental stages. PLoS ONE 2016, 11, e0161667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Cai, Y.; Zhuo, Z.; Yang, W.; Yang, C.; Zhang, J. Transcriptome analysis in different developmental stages of Batocera horsfieldi (Coleoptera: Cerambycidae) and comparison of candidate olfactory genes. PLoS ONE 2018, 13, e0192730. [Google Scholar] [CrossRef] [Green Version]
- Noriega, D.D.; Arias, P.L.; Barbosa, H.R.; Arraes, F.B.M.; Ossa, G.A.; Villegas, B.; Coelho, R.R.; Albuquerque, E.V.S.; Togawa, R.C.; Grynberg, P.; et al. Transcriptome and gene expression analysis of three developmental stages of the coffee berry borer, Hypothenemus hampei. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Lu, C.; Geib, S.M.; Zheng, J.; Wu, S.; Zhang, F.; Liang, G. Characterization of Dendrolimus houi Lajonquiere (Lepidoptera: Lasiocampidae) transcriptome across all life stages. Insects 2019, 10, 442. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, C.G.C.; Spaink, H.P.; Van Der Zee, M. The extraembryonic serosa is a frontier epithelium providing the insect egg with a full-range innate immune response. Elife 2014, 3, 1–21. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, J.; Liu, Q.; Zhang, Y.; Gong, J.; Hou, Y. Genome-wide analysis and hormone regulation of chitin deacetylases in silkworm. Int. J. Mol. Sci. 2019, 20, 1679. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-F.; Mu, L.-L.; Guo, W.-C.; Li, G.-Q. Identification and hormone induction of putative chitin synthase genes and splice variants in Leptinotarsa decemlineata (Say). Arch. Insect Biochem. Physiol. 2016, 92, 242–258. [Google Scholar] [CrossRef]
- Okamoto, N.; Yamanaka, N.; Satake, H.; Saegusa, H.; Kataoka, H.; Mizoguchi, A. An ecdysteroid-inducible insulin-like growth factor-like peptide regulates adult development of the silkmoth Bombyx mori. FEBS J. 2009, 276, 1221–1232. [Google Scholar] [CrossRef]
- Dong, Y.; Desneux, N.; Lei, C.; Niu, C. Transcriptome characterization analysis of Bactrocera minax and new insights into its pupal diapause development with gene expression analysis. Int. J. Biol. Sci. 2014, 10, 1051–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakeel, M.; Du, J.; Li, S.W.; Zhou, Y.J.; Sarwar, N.; Bukhari, S.A.H. Characterization, knockdown and parental effect of hexokinase gene of Cnaphalocrocis medinalis (Lepidoptera: Pyralidae) revealed by RNA interference. Genes (Basel) 2020, 11, 1258. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, E.; Wang, L.; Margo, G. Synthesis of chitin in cell-free extracts of Prodenia eridania. Nature 1963, 198, 790. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, M.; Shen, Q.; Liu, X.; Shi, Z.; Wang, S.; Tang, B. Functional characterization of three trehalase genes regulating the chitin metabolism pathway in rice brown planthopper using RNA interference. Sci. Rep. 2016, 6, 1–14. [Google Scholar]
- Tang, B.; Wei, P.; Zhao, L.; Shi, Z.; Shen, Q.; Yang, M.; Xie, G.; Wang, S. Knockdown of five trehalase genes using RNA interference regulates the gene expression of the chitin biosynthesis pathway in Tribolium castaneum. BMC Biotechnol. 2016, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Tsuji, N.; Miyoshi, T.; Motobu, M.; Islam, M.K.; Alim, M.A.; Fujisaki, K. Characterization of glutamine: Fructose-6-phosphate aminotransferase from the ixodid tick, Haemaphysalis longicornis, and its critical role in host blood feeding. Int. J. Parasitol. 2007, 37, 383–392. [Google Scholar] [CrossRef]
- Freedman, A.H.; Clamp, M.; Sackton, T.B. Error, noise and bias in de novo transcriptome assemblies. Mol. Ecol. Resour. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Foster, J.M.; Nelson, L.S.; Ma, D.; Carlow, C.K.S. The chitin synthase genes chs-1 and chs-2 are essential for C. elegans development and responsible for chitin deposition in the eggshell and pharynx, respectively. Dev. Biol. 2005, 285, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Kono, N.; Nakamura, H.; Ohtoshi, R.; Tomita, M.; Numata, K.; Arakawa, K. The bagworm genome reveals a unique fibroin gene that provides high tensile strength. Commun. Biol. 2019, 2, 1–9. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, M.; Pandher, S.; Kaur, G.; Goel, N.; Rathore, P. Using de novo transcriptome assembly and analysis to study RNAi in Phenacoccus solenopsis Tinsley (Hemiptera: Pseudococcidae). Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef]
- Senthil Kumar, R.; Srinivasan, R.; Rawdzah, M.A.; Malini, P. Mapping and identification of potential target genes from short–RNA Seq for the control of Pieris rapae larvae. Genomics 2020, 112, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.K.; Nagaraju, J.; Tomar, A.; Bentur, J.S.; Nair, S. Pyrosequencing-based transcriptome analysis of the asian rice gall midge reveals differential response during compatible and incompatible interaction. Int. J. Mol. Sci. 2012, 13, 13079–13103. [Google Scholar] [CrossRef]
- Ding, S.; Wang, S.; He, K.; Jiang, M.; Li, F. Large-scale analysis reveals that the genome features of simple sequence repeats are generally conserved at the family level in insects. BMC Genom. 2017, 18, 1–10. [Google Scholar] [CrossRef]
- Acquadro, A.; Torello Marinoni, D.; Sartor, C.; Dini, F.; Macchio, M.; Botta, R. Transcriptome characterization and expression profiling in chestnut cultivars resistant or susceptible to the gall wasp Dryocosmus kuriphilus. Mol. Genet. Genom. 2020, 295, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Lokki, J.; Suomalainen, E.; Saura, A.; Lankinen, P. Genetic polymorphism and evolution in parthenogenetic animals. ii. diploid and polyploid Solenobia triquetrella (Lepidoptera: Psychidae). Genetics 1975, 79, 513–525. [Google Scholar] [PubMed]
- Shao, Z.; Li, Y.; Ding, J.; Liu, Z.; Zhang, X. Identification, characterization, and functional analysis of chitin synthase genes in Glyphodes pyloalis Walker (Lepidoptera: Pyralidae). Int. J. Mol. Sci. 2020, 21, 4656. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.J.; Wu, Y.B.; Chen, L.; Xu, K.K.; Xie, Y.F.; Wang, J.J. Two chitin biosynthesis pathway genes in Bactrocera dorsalis (Diptera: Tephritidae): Molecular characteristics, expression patterns, and roles in larval-pupal transition. J. Econ. Entomol. 2015, 108, 2433–2442. [Google Scholar] [CrossRef]
- Bubici, G.; Prigigallo, M.I.; Garganese, F.; Nugnes, F.; Jansen, M.; Porcelli, F. First report of Aleurocanthus spiniferus on Ailanthus altissima: Profiling of the insect microbiome and MicroRNAs. Insects 2020, 11, 161. [Google Scholar] [CrossRef] [Green Version]
- Douglas, A.E. Nutritional interactions in insect-microbial symbioses: Aphids and their symbiotic bacteria buchnera. Annu. Rev. Entomol. 1998, 43, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Haine, E.R. Symbiont-mediated protection. Proc. R. Soc. B Biol. Sci. 2008, 275, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlie, J.C.; Johnson, K.N. Symbiont-mediated protection in insect hosts. Trends Microbiol. 2009, 17, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Engelstädter, J.; Hurst, G.D. The ecology and evolution of microbes that manipulate host reproduction. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 127–149. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Statistics of the Raw Reads | ||
|---|---|---|
| No. of base pairs before trimming (bp) | 67,119,034,800 | |
| No. of base pairs after trimming (bp) | 67,068,586,489 | |
| No. of clean reads | 223,730,116 | |
| Q30 | 92.565% | |
| GC content | 44.44% | |
| Data statistics of M. plana’s transcriptome | ||
| Type | Initial Assembly | Final Assembly |
| Total numbers of transcripts | 330,017 | 193,686 |
| Completed BUSCOs | 97.95% | 97.95% |
| Total length (bp) | 296,149,848 | 175,630,488 |
| N50 (bp) | 1785 | 1981 |
| Average length (bp) | 897.38 | 906.78 |
| Max length (bp) | 59,444 | 59,444 |
| Min length (bp) | 176 | 182 |
| GC content | 38.47% | 39.46 |
| Length distribution of final transcriptome assembly (bp) | ||
| 200 | 28 | |
| 300 | 64,955 | |
| 500 | 54,967 | |
| 1000 | 28,633 | |
| >2000 | 21,826 | |
| Functional annotation statistics | ||
| Total transcripts | 193,686 | |
| SwissProt and NR | 59,571 | |
| TrEMBL | 56,704 | |
| GO | 46,534 | |
| KOG | 50,246 | |
| KEGG | 1012 | |
| Chitin Biosynthesis Genes | Comparative Expression Analysis | |||||
|---|---|---|---|---|---|---|
| Egg vs. Larva | Larva vs. Pupa | Pupa vs. Adult | ||||
| Down-Regulated | Up-Regulated | Down-Regulated | Up-Regulated | Down-Regulated | Up-Regulated | |
| Trehalase | M. plana, B. mori [5] | M. plana, B. mori [5] | ||||
| Hexokinase | M. plana | B. mori, D. punctatus [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] | M. plana, B. mori [5] | D. punctatus [49] | M. plana | |
| Glucose-6-phosphate isomerase | M. plana | D. punctatus [49] | B. mori [5] | D. punctatus [49] | ||
| Glutamine:fructose-6-phosphate aminotransferase | D. puctatus [49] | M. plana | ||||
| Glucosamine-6-phosphate N-acetyltransferase | B. mori [5] | |||||
| Phosphoacetyl-glucosamine mutase | B. mori, D. punctatus [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] | D. punctatus [49] | ||||
| UDP-N-acetyl-glucosamine pyrophosphorylase | B. mori, D. punctatus [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48] | D. punctatus [49] | D. punctatus [49] | B. horsfieldi [50] | ||
| Chitin synthase | D. punctatus [49] | M. plana | M. plana, D. punctatus [49] | B. mori [5] | D. punctatus [49] | M. plana, B. horsfieldi,H. hampei [35,36,37,38,39,40,41,42,43,44,45,46,47,48,49] |
| Microsatellite (SSR) Sequences Distribution | |||
|---|---|---|---|
| SSR Type | Repeat Motif | Number | Frequency (%) |
| Di-nucleotide | AT/GC/AG/CT Other Types | 17,015 | 79.08 |
| Tri-nucleotide | CGC/TTA/TGA/AAC Other Types | 3956 | 18.39 |
| Tetra-nucleotide | TGTC/GACA/ACAG/GCAC Other Types | 424 | 1.97 |
| Penta-nucleotide | CGAGA/ATTTG/CATTC Other Types | 46 | 0.21 |
| Hexa-nucleotide | CGCGCC/GGGCGC/TTTGAA Other Types | 32 | 0.15 |
| >6 nucleotide | GCGGGCG/GGCGGGC Other Types | 43 | 0.20 |
| Total | 21,516 | 100 | |
| Statistic of SNP Variants | |||
| Sample | HomoSNP | HeteroSNP | All |
| Egg | 44,650 | 57,742 | 102,392 |
| Larva | 31,708 | 47,606 | 79,314 |
| Pupa | 42,711 | 58,859 | 101,570 |
| Adult | 40,687 | 55,932 | 96,619 |
| Total | 159,756 | 220,139 | 379,895 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmat, N.L.; Zifruddin, A.N.; Zainal Abidin, C.M.R.; Nor Muhammad, N.-A.; Hassan, M. The Developmental Transcriptome of Bagworm, Metisa plana (Lepidoptera: Psychidae) and Insights into Chitin Biosynthesis Genes. Genes 2021, 12, 7. https://doi.org/10.3390/genes12010007
Rahmat NL, Zifruddin AN, Zainal Abidin CMR, Nor Muhammad N-A, Hassan M. The Developmental Transcriptome of Bagworm, Metisa plana (Lepidoptera: Psychidae) and Insights into Chitin Biosynthesis Genes. Genes. 2021; 12(1):7. https://doi.org/10.3390/genes12010007
Chicago/Turabian StyleRahmat, Nur Lina, Anis Nadyra Zifruddin, Cik Mohd Rizuan Zainal Abidin, Nor-Azlan Nor Muhammad, and Maizom Hassan. 2021. "The Developmental Transcriptome of Bagworm, Metisa plana (Lepidoptera: Psychidae) and Insights into Chitin Biosynthesis Genes" Genes 12, no. 1: 7. https://doi.org/10.3390/genes12010007