Sequencing Red Fox Y Chromosome Fragments to Develop Phylogenetically Informative SNP Markers and Glimpse Male-Specific Trans-Pacific Phylogeography

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
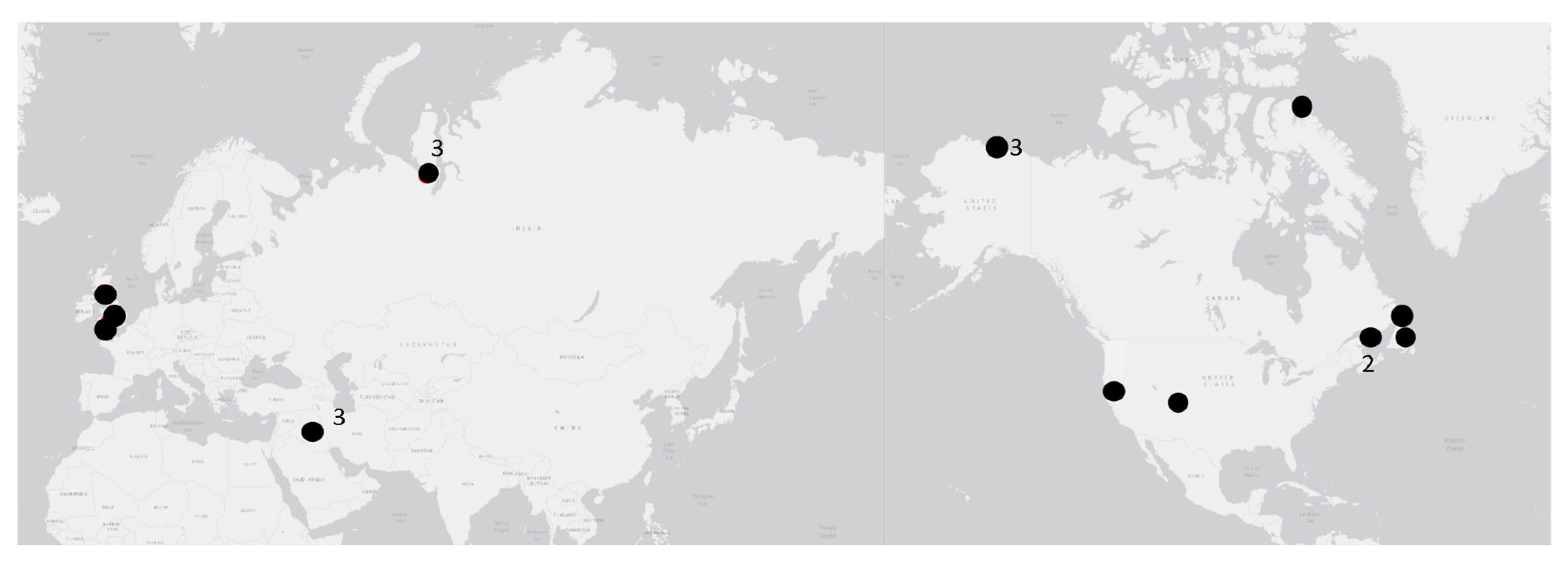
2.1. Samples
2.2. Capture Enrichment and Sequencing
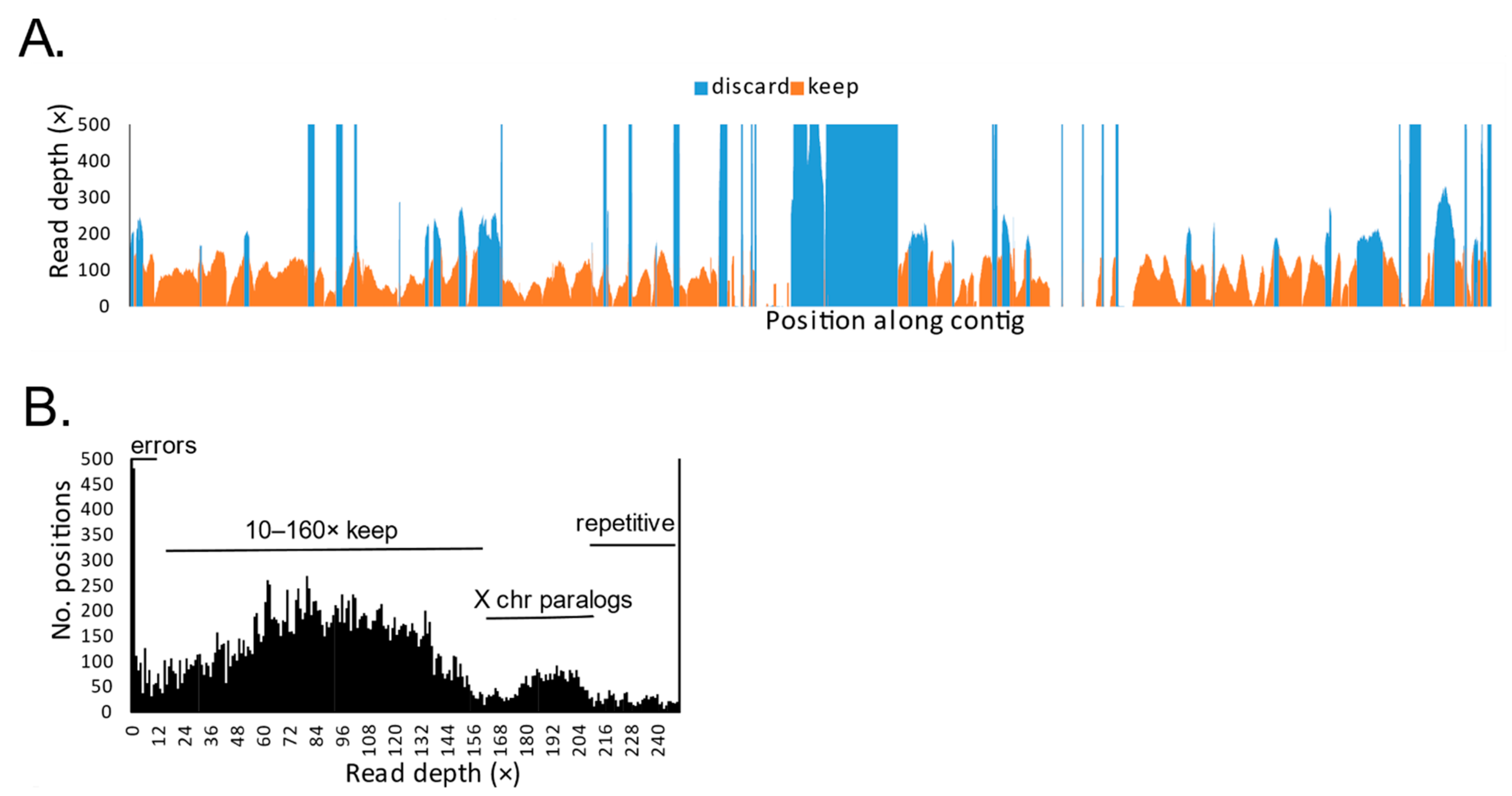
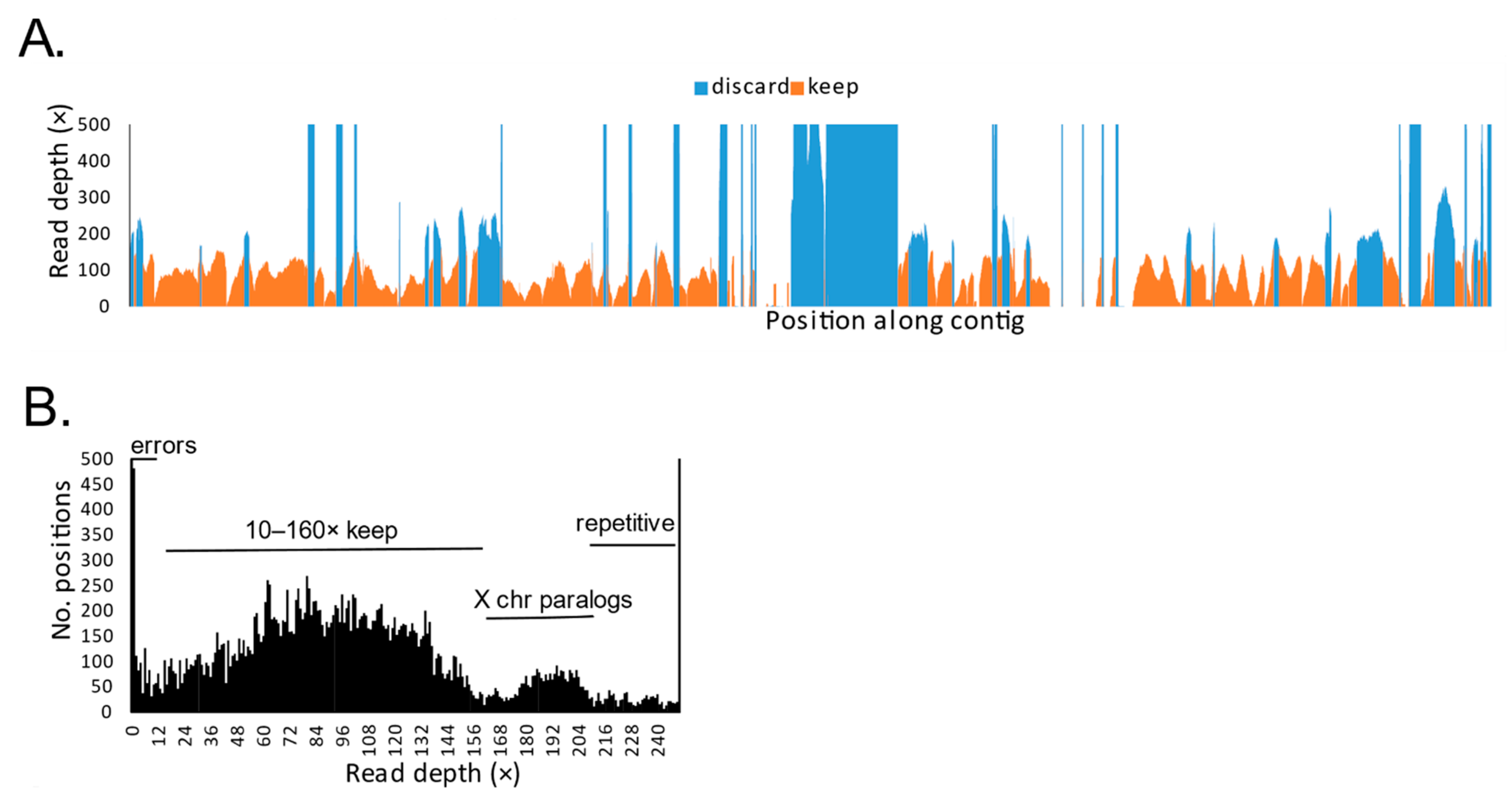
2.3. Bioinformatic Processing
2.4. Phylogenetic Analysis
2.5. SNP Assay
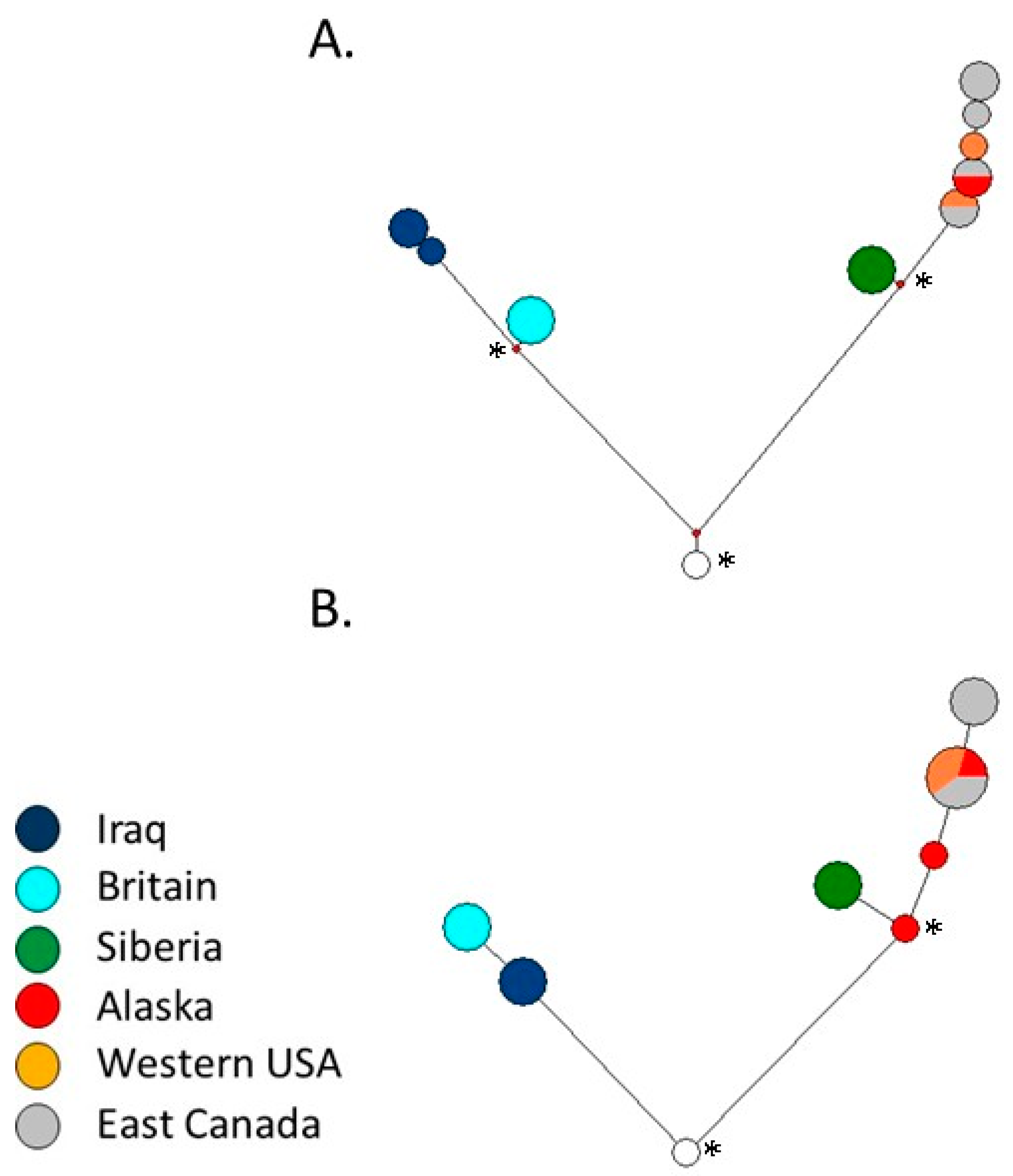
2.6. Intraspecific Red Fox Network
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, C.J.; Soulsbury, C.D.; Statham, M.J.; Ho, S.Y.; Wall, D.; Dolf, G.; Iossa, G.; Baker, P.J.; Harris, S.; Sacks, B.N.; et al. Temporal genetic variation of the red fox, Vulpes vulpes, across western Europe and the British Isles. Quat. Sci. Rev. 2012, 57, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubry, K.B.; Statham, M.J.; Sacks, B.N.; Perrine, J.D.; Wisely, S.M. Phylogeography of the North American red fox: Vicariance in Pleistocene forest refugia. Mol. Ecol. 2009, 18, 2668–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutschera, V.E.; LeComte, N.; Janke, A.; Selva, N.; Sokolov, A.A.; Haun, T.; Steyer, K.; Nowak, C.; Hailer, F. A range-wide synthesis and timeline for phylogeographic events in the red fox (Vulpes vulpes). BMC Evol. Biol. 2013, 13, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statham, M.J.; Murdoch, J.; Janecka, J.; Aubry, K.B.; Edwards, C.J.; Soulsbury, C.D.; Berry, O.; Wang, Z.; Harrison, D.; Pearch, M.; et al. Range-wide multilocus phylogeography of the red fox reveals ancient continental divergence, minimal genomic exchange and distinct demographic histories. Mol. Ecol. 2014, 23, 4813–4830. [Google Scholar] [CrossRef] [PubMed]
- Statham, M.J.; Edwards, C.; Norén, K.; Soulsbury, C.; Sacks, B.N. Genetic analysis of European red foxes reveals multiple distinct peripheral populations and central continental admixture. Quat. Sci. Rev. 2018, 197, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, E.W.; Renshaw, B.; Clement, C.J.; Himschoot, E.A.; Hundertmark, K.J.; Hueffer, K. Population structure of two rabies hosts relative to the known distribution of rabies virus variants in Alaska. Mol. Ecol. 2016, 25, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Sacks, B.N.; Lounsberry, Z.T.; Statham, M. Nuclear Genetic Analysis of the Red Fox Across its Trans-Pacific Range. J. Hered. 2018, 109, 573–584. [Google Scholar] [CrossRef]
- Rando, H.M.; Stutchman, J.T.; Bastounes, E.R.; Johnson, J.L.; Driscoll, C.A.; Barr, C.S.; Trut, L.N.; Sacks, B.N.; Kukekova, A.V. Y-Chromosome Markers for the Red Fox. J. Hered. 2017, 108, 678–685. [Google Scholar] [CrossRef]
- Skaletsky, H.; Kuroda-Kawaguchi, T.; Minx, P.J.; Cordum, H.S.; Hillier, L.; Brown, L.G.; Repping, S.; Pyntikova, T.; Ali, J.; Bieri, T.; et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 2003, 423, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Tomaszkiewicz, M.; Rangavittal, S.; Cechova, M.; Sanchez, R.C.; Fescemyer, H.W.; Harris, R.; Ye, D.; O’Brien, P.C.; Chikhi, R.; Ryder, O.A.; et al. A time- and cost-effective strategy to sequence mammalian Y Chromosomes: An application to the de novo assembly of gorilla Y. Genome Res. 2016, 26, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Charlesworth, D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. B Biol. Sci. 2000, 355, 1563–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangavittal, S.; Harris, R.S.; Cechova, M.; Tomaszkiewicz, M.; Chikhi, R.; Makova, K.D.; Medvedev, P. RecoverY: K-mer-based read classification for Y-chromosome-specific sequencing and assembly. Bioinformatics 2017, 34, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natanaelsson, C.; Oskarsson, M.C.; Angleby, H.; Lundeberg, J.; Kirkness, E.; Savolainen, P. Dog Y chromosomal DNA sequence: Identification, sequencing and SNP discovery. BMC Genet. 2006, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Davis, B.W.; Raudsepp, T.; Wilkerson, A.J.P.; Mason, V.C.; Ferguson-Smith, M.; O’Brien, P.C.; Waters, P.D.; Murphy, W.J. Comparative analysis of mammalian Y chromosomes illuminates ancestral structure and lineage-specific evolution. Genome Res. 2013, 23, 1486–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellborg, L.; Ellegren, H. Y chromosome conserved anchored tagged sequences (YCATS) for the analysis of mammalian male-specific DNA. Mol. Ecol. 2002, 12, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.-L.; Oskarsson, M.; Ardalan, A.; Angleby, H.; Dahlgren, L.-G.; Tepeli, C.; Kirkness, E.; Savolainen, P.; Zhang, Y.E. Origins of domestic dog in Southern East Asia is supported by analysis of Y-chromosome DNA. Heredity 2011, 108, 507–514. [Google Scholar] [CrossRef]
- Tsubouchi, A.; Fukui, D.; Ueda, M.; Tada, K.; Toyoshima, S.; Takami, K.; Tsujimoto, T.; Uraguchi, K.; Raichev, E.; Kaneko, Y.; et al. Comparative molecular phylogeny and evolution of sex chromosome DNA sequences in the family Canidae (Mammalia: Carnivora). Zool. Sci. 2012, 29, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Rando, H.M.; Wadlington, W.H.; Johnson, J.L.; Stutchman, J.T.; Trut, L.N.; Farré, M.; Kukekova, A.V. The Red Fox Y-Chromosome in Comparative Context. Genes 2019, 10, 409. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Khaleel, S.S.; Huang, H.; Wu, C.H. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol. Med. 2014, 9, 8. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Project, G.; et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Wayne, R.K.; Geffen, E.; Girman, D.J.; Koepfli, K.P.; Lau, L.M.; Marshall, C.R. Molecular systematics of the Canidae. Syst. Biol. 1997, 46, 622–653. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., III; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Perini, F.A.; Russo, C.A.M.; Schrago, C.G. The evolution of South American endemic canids: A history of rapid diversification and morphological parallelism. J. Evol. Biol. 2010, 23, 311–322. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.-J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Forster, P.; Röhl, A.; Lünnemann, P.; Brinkmann, C.; Zerjal, T.; Tyler-Smith, C.; Brinkmann, B. A short tandem repeat-based phylogeny for the human Y chromosome. Am. J. Hum. Genet. 2000, 67, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Saillard, J.; Forster, P.; Lynnerup, N.; Bandelt, H.-J.; Nørby, S. mtDNA Variation among Greenland Eskimos: The edge of the Beringian expansion. Am. J. Hum. Genet. 2000, 67, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Long, J.L. Introduced Mammals of the World: Their History, Distribution and Influence; CABI Publishing: Wallingford, UK, 2003; 589p. [Google Scholar]
- Kasprowicz, A.E.; Statham, M.J.; Sacks, B.N. Fate of the other redcoat: Remnants of colonial British foxes in the eastern United States. J. Mammal. 2015, 97, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Sacks, B.N.; Brown, S.K.; Stephens, D.; Pedersen, N.C.; Wu, J.-T.; Berry, O. Y chromosome analysis of dingoes and Southeast Asian village dogs suggests a Neolithic continental expansion from Southeast Asia followed by multiple Austronesian dispersals. Mol. Biol. Evol. 2013, 30, 1103–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Sample Identifier | Single Nucleotide Polymorphisms of 31 Y Chromosome Loci a |
|---|---|---|
| Ancestral node | G C C C C T T C T A G C A A G T T T G C A C T C A C T T A C G | |
| Britain | S08-0428 | C T - - - - C - - T - - - - A - - A - A C A C - - - - - - - - |
| Britain | S08-0433 | C T - - - - C - - T - - - - A - - A - A C A C - - - - - - - - |
| Britain | S08-0436 | C T - - - - C - - T - - - - A - - A - A C A C - - - - - - - - |
| Iraq | S10-0036 * | C - - - - - C - - T - - - G A C - A - A C A C - - T - - - T A |
| Iraq | S10-0039 | C - - - - - C - - T - - - G A C - A - A C A C - - T - - - T A |
| Iraq | S14-1369 | C - - - - - C - - T - - - G A C - A - A C A C - - - - - - T A |
| Yamal, Siberia | S12-0237 * | - - A T T C - T G - - - - - - - - - A - C - - - - T G G G - - |
| Yamal, Siberia | S12-0241 * | - - A T T C - T G - - - - - - - - - A - C - - - - T G G G - - |
| Yamal, Siberia | S12-0244 * | - - A T T C - T G - - - - - - - - - A - C - - - - T G G G - - |
| Alaska, USA | S12-1162 | - - A T T C - - G - C T - - - - C - A - C - - T - T G G G - - |
| Bylott Island, Canada | S12-1600 * | - - A T T C - - G - C T T - - - C - A - C - - T G T G G G - - |
| Eastern Canada | S12-1607 * | - - A T T C - - G - C T - - - - C - A - C - - T - T G G G - - |
| Eastern Canada | S12-1608 * | - - A T T C - - G - C T - - - - C - A - C - - - - T G G G - - |
| Eastern Canada | S14-0418 | - - A T T C - - G - C T T - - - C - A - - - - T G T G G G - - |
| Eastern Canada | S14-0432 | - - A T T C - - G - C T T - - - C - A - - - - T G T G G G - - |
| Lassen Co, CA, USA | M1 | - - A T T C - - G - C T - - - - C - A - C - - - - T G G G - - |
| Gunnison, CO, USA | S12-1176 * | - - A T T C - - G - C T - - - - C - A - C - - T G T G G G - - |
| Alaska, USA | S12-1163 * | - - A T T C - - G - - - - X X X X X X X X X X X X X X X X X X |
| Alaska, USA | S12-1161 ** | - - A T T C - - G - C - - X X X X X X X X X X X X X X X X X X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sacks, B.N.; Lounsberry, Z.T.; Rando, H.M.; Kluepfel, K.; Fain, S.R.; Brown, S.K.; Kukekova, A.V. Sequencing Red Fox Y Chromosome Fragments to Develop Phylogenetically Informative SNP Markers and Glimpse Male-Specific Trans-Pacific Phylogeography. Genes 2021, 12, 97. https://doi.org/10.3390/genes12010097
Sacks BN, Lounsberry ZT, Rando HM, Kluepfel K, Fain SR, Brown SK, Kukekova AV. Sequencing Red Fox Y Chromosome Fragments to Develop Phylogenetically Informative SNP Markers and Glimpse Male-Specific Trans-Pacific Phylogeography. Genes. 2021; 12(1):97. https://doi.org/10.3390/genes12010097
Chicago/Turabian StyleSacks, Benjamin N., Zachary T. Lounsberry, Halie M. Rando, Kristopher Kluepfel, Steven R. Fain, Sarah K. Brown, and Anna V. Kukekova. 2021. "Sequencing Red Fox Y Chromosome Fragments to Develop Phylogenetically Informative SNP Markers and Glimpse Male-Specific Trans-Pacific Phylogeography" Genes 12, no. 1: 97. https://doi.org/10.3390/genes12010097
APA StyleSacks, B. N., Lounsberry, Z. T., Rando, H. M., Kluepfel, K., Fain, S. R., Brown, S. K., & Kukekova, A. V. (2021). Sequencing Red Fox Y Chromosome Fragments to Develop Phylogenetically Informative SNP Markers and Glimpse Male-Specific Trans-Pacific Phylogeography. Genes, 12(1), 97. https://doi.org/10.3390/genes12010097