
Transcriptome Analysis of In Vitro Fertilization and Parthenogenesis Activation during Early Embryonic Development in Pigs


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Ethics Statement
2.2. Collection and Culture of Pig Oocytes and Embryos
2.2.1. Oocyte Collection
2.2.2. In Vitro Fertilization
2.2.3. Parthenogenetic Activation
2.3. Preparation of cDNA and Transcription Profiling
2.4. Quality Control (QC) and Transcriptome Assembly
2.5. Expression Calculation and Differentially Expressed Analysis
2.6. Identification and Function Analysis of lincRNAs
2.6.1. Neighboring Gene Analysis
2.6.2. Weighted Gene Co-Expression Network Analysis
2.6.3. Identification of Hub LincRNAs
2.7. Gene Ontology and Pathway Analysis
2.8. Validation of RNA-Seq Data by Quantitative Reverse-Transcription PCR
3. Results
3.1. Overview of Sequencing Data
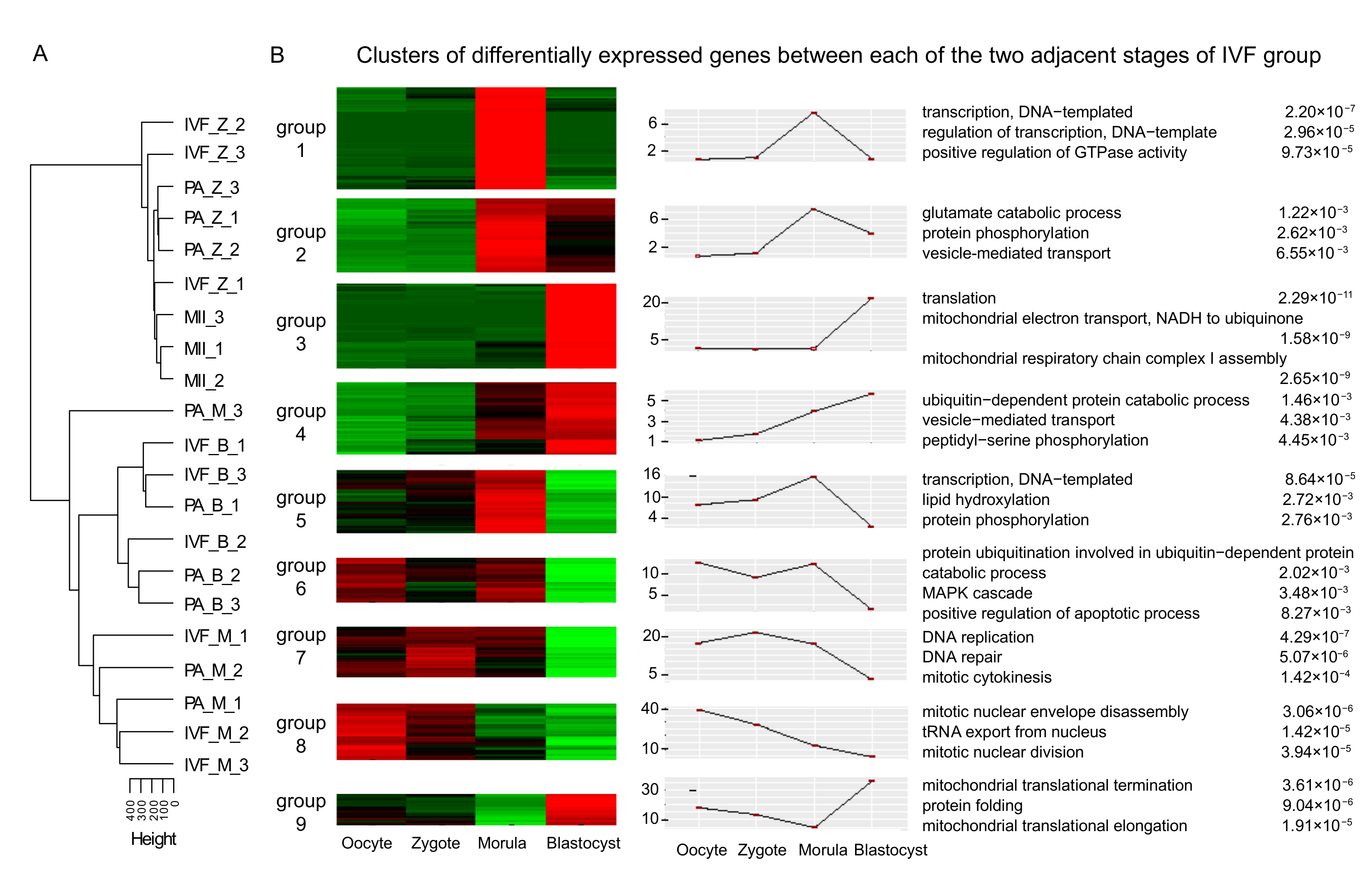
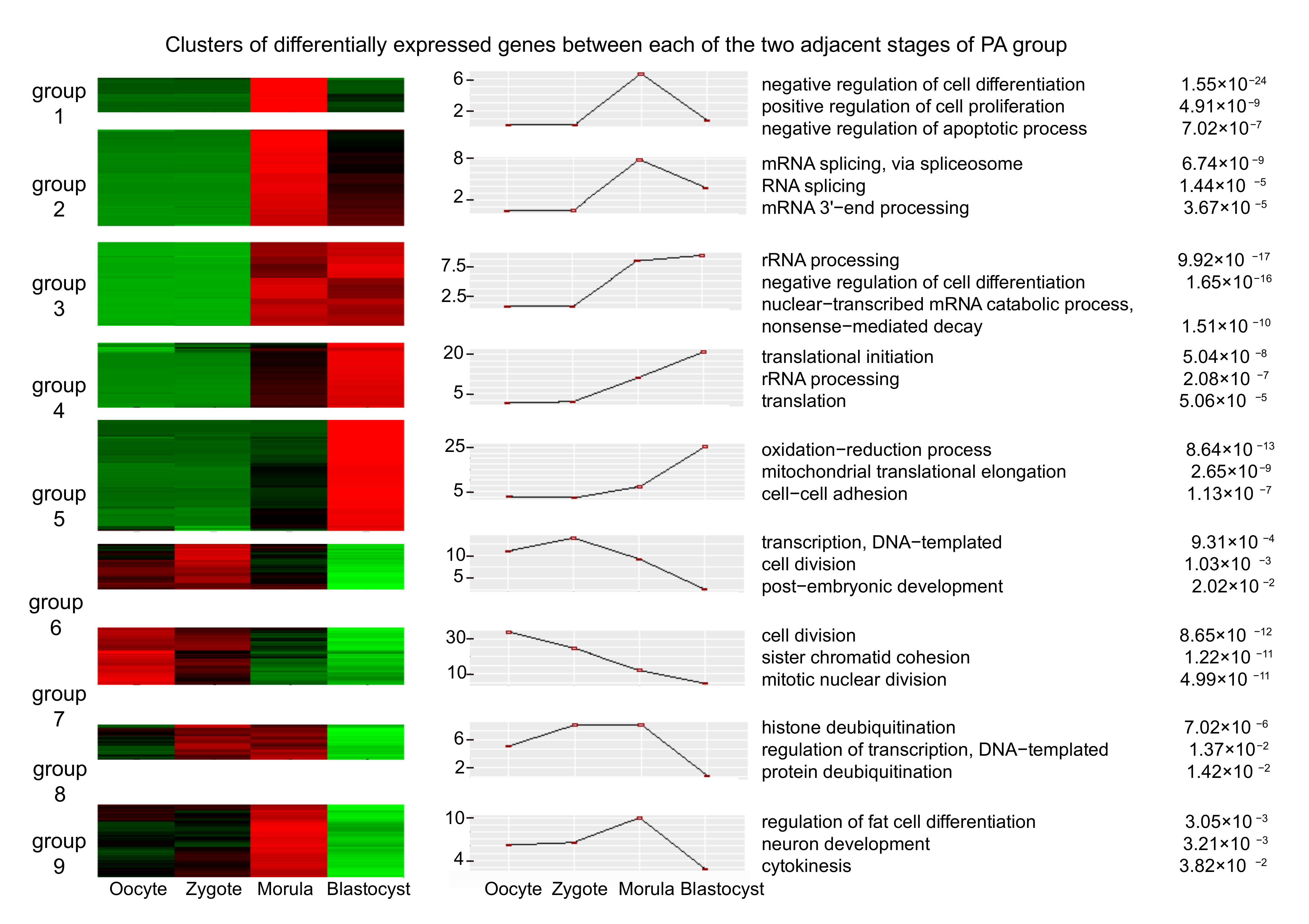
3.2. Differential Expression Analysis of mRNAs in Adjacent Stages, IVF, and PA
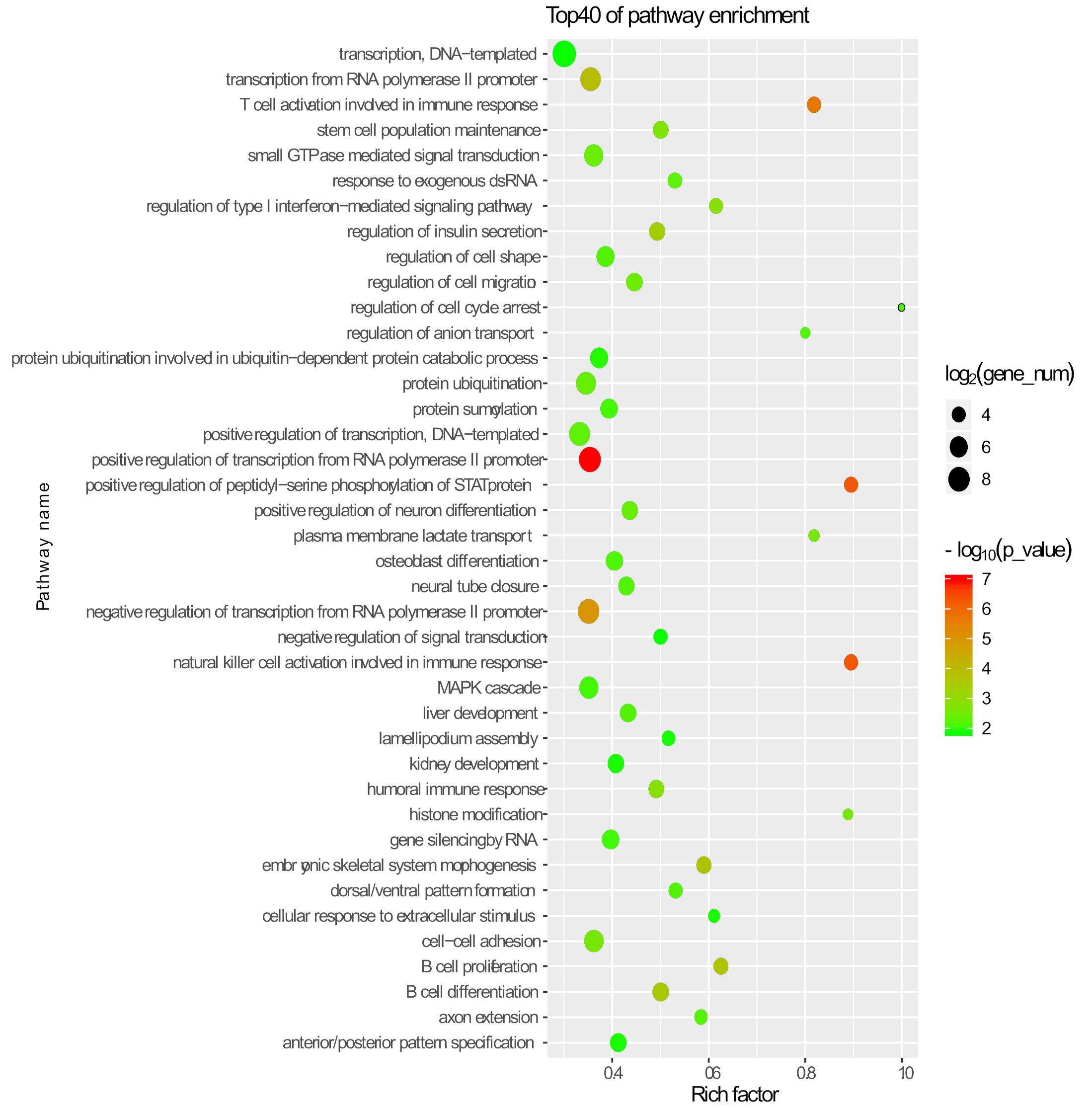
3.3. Identification and Function Prediction Analysis of lincRNAs
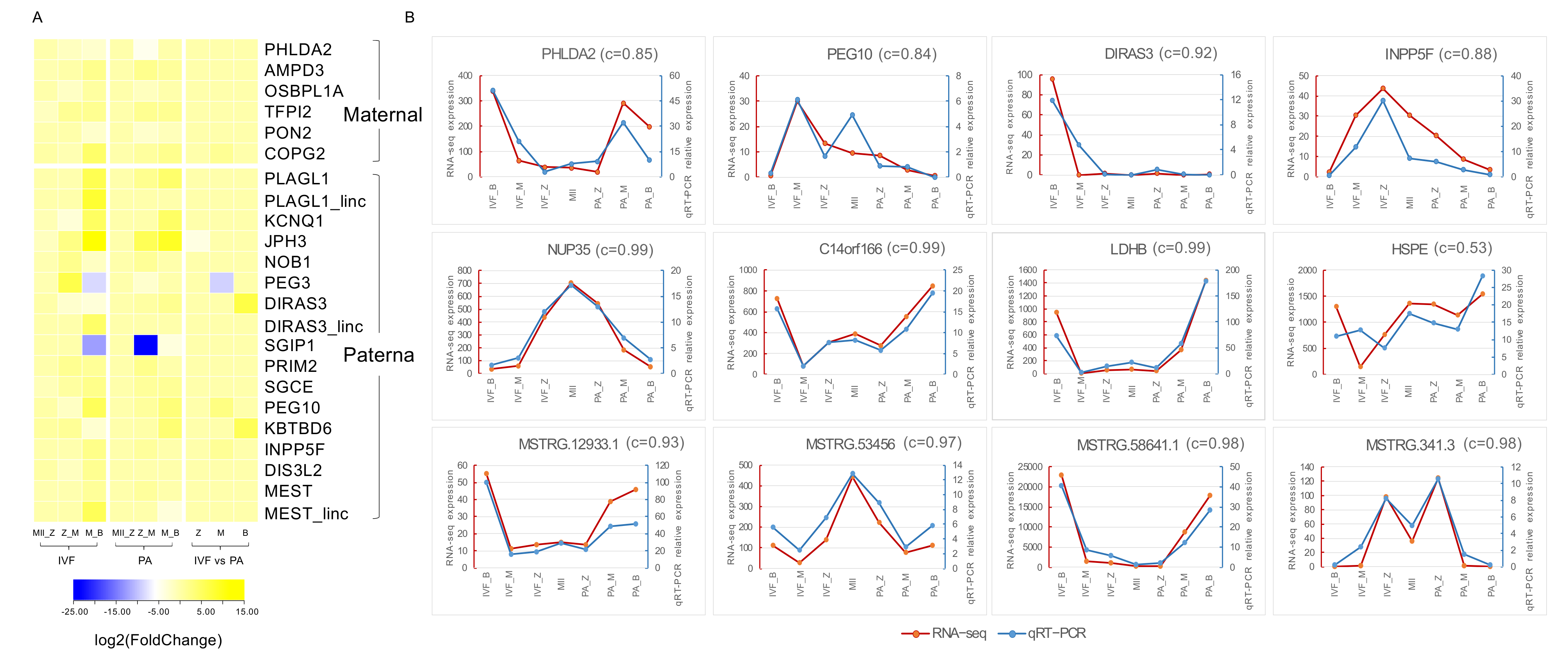
3.4. Identification of Imprinted Genes in Pig
3.5. Quantitative Reverse-Transcription PCR Validation of Transcriptome Sequencing Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Brevini, T.; Antonini, S.; Cillo, F.; Crestan, M.; Gandolfi, F. Porcine embryonic stem cells: Facts, challenges and hopes. Theriogenology 2007, 68, S206–S213. [Google Scholar] [CrossRef]
- Lunney, J.K. Advances in Swine Biomedical Model Genomics. Int. J. Biol. Sci. 2007, 3, 179–184. [Google Scholar] [CrossRef]
- Hall, V. Porcine Embryonic Stem Cells: A Possible Source for Cell Replacement Therapy. Stem Cell Rev. Rep. 2008, 4, 275–282. [Google Scholar] [CrossRef]
- Patterson, J.K.; Lei, X.G.; Miller, D.D. The Pig as an Experimental Model for Elucidating the Mechanisms Governing Dietary Influence on Mineral Absorption. Exp. Biol. Med. 2008, 233, 651–664. [Google Scholar] [CrossRef]
- Dua, D.; Nagoorvali, D.; Chauhan, M.S.; Palta, P.; Mathur, P.; Singh, M.K. Calcium ionophore enhanced developmental competence and apoptotic dynamics of goat parthenogenetic embryos produced in vitro. Vitr. Cell. Dev. Biol. Anim. 2019, 55, 159–168. [Google Scholar] [CrossRef]
- Cibelli, J.B.; Cunniff, K.; Vrana, K.E. Embryonic Stem Cells from Parthenotes. Methods Enzym. 2006, 418, 117–135. [Google Scholar] [CrossRef]
- Brevini, T.A.; Gandolfi, F. Parthenotes as a source of embryonic stem cells. Cell Prolif. 2007, 41, 20–30. [Google Scholar] [CrossRef]
- Kono, T.; Obata, Y.; Wu, Q.; Niwa, K.; Ono, Y.; Yamamoto, Y.; Park, E.S.; Seo, J.-S.; Ogawa, H. Birth of parthenogenetic mice that can develop to adulthood. Nature 2004, 428, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; King, T.; Dobrinsky, J.; Harkness, L.; Ferrier, T.; Bosma, W.; Schreier, L.L.; Guthrie, H.D.; DeSousa, P.; Wilmut, I. In Vitro and In Vivo Developmental Competence of Ovulated andIn Vitro Matured Porcine Oocytes Activated by Electrical Activation. Cloning Stem Cells 2003, 5, 355–365. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Barton, S.C.; Surani, M.A.H.; Norris, M.L. Role of paternal and maternal genomes in mouse development. Nature 1984, 311, 374–376. [Google Scholar] [CrossRef]
- Tada, T.; Takagi, N. Early development and X-chromosome inactivation in mouse parthenogenetic embryos. Mol. Reprod. Dev. 1992, 31, 20–27. [Google Scholar] [CrossRef]
- Deshmukh, R.S.; Østrup, O.; Strejcek, F.; Vejlsted, M.; Lucas-Hahn, A.; Petersen, B.; Li, J.; Callesen, H.; Niemann, H.; Hyttel, P. Early Aberrations in Chromatin Dynamics in Embryos Produced Under In Vitro Conditions. Cell. Reprogr. 2012, 14, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.B.; Xuan, M.F.; Han, S.Z.; Li, Z.Y.; Khan, N.; Quan, B.H.; Yin, X.J.; Kang, J.D. Ginsenoside Rb1 protects porcine oocytes against methylglyoxal damage thus it improves the quality of parthenogenetic activation and in vitro fertilization embryos. Environ. Toxicol. 2020, 36, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.T.K.; Nguyen, T.V.T.; Van Nguyen, T.; Bui, H.-T. Epigenetic impairments in development of parthenogenetic preimplantation mouse embryos. J. Reprod. Dev. 2019, 65, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Fu, Q.; Pu, L.; Zhang, P.; Huang, Y.; Hou, Z.; Xu, Z.; Chen, D.; Huang, F.; Deng, T.; et al. Integrated Analysis of Quantitative Proteome and Transcriptional Profiles Reveals the Dynamic Function of Maternally Expressed Proteins After Parthenogenetic Activation of Buffalo Oocyte. Mol. Cell. Proteom. 2018, 17, 1875–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isom, S.C.; Stevens, J.R.; Li, R.; Spollen, W.G.; Cox, L.; Spate, L.D.; Murphy, C.N.; Prather, R.S. Transcriptional profiling by RNA-Seq of peri-attachment porcine embryos generated by a variety of assisted reproductive technologies. Physiol. Genom. 2013, 45, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-P.; Duan, F.-F.; Zhao, Y.-T.; Gu, K.-L.; Liao, L.-Q.; Su, H.-B.; Hao, J.; Zhang, K.; Yang, N.; Wang, Y. A TRIM71 binding long noncoding RNA Trincr1 represses FGF/ERK signaling in embryonic stem cells. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xu, H.; Li, T.; Wu, J.; An, L.; Zhao, Z.; Xiao, M.; Adu-Asiamah, P.; Zhang, X.; Zhang, L. Chicken GHR antisense transcript regulates its sense transcript in hepatocytes. Gene 2018, 682, 101–110. [Google Scholar] [CrossRef]
- Blank-Giwojna, A.; Postepska-Igielska, A.; Grummt, I. lncRNA KHPS1 Activates a Poised Enhancer by Triplex-Dependent Recruitment of Epigenomic Regulators. Cell Rep. 2019, 26, 2904–2915.e4. [Google Scholar] [CrossRef] [Green Version]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gao, Z.; Wang, X.; Liu, H.; Zhang, Y.; Liu, Z. Identification and functional analysis of long intergenic noncoding RNA genes in porcine pre-implantation embryonic development. Sci. Rep. 2016, 6, 38333. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Liu, Y.; Wu, Q.; Li, D.; Zhang, L.; Wu, X.; Wang, R.; Zhang, D.; Gao, S.; Li, W. Long non-coding RNAs potentially function synergistically in the cellular reprogramming of SCNT embryos. BMC Genom. 2018, 19, 631. [Google Scholar] [CrossRef]
- Kuang, L.; Lei, M.; Li, C.; Guo, Z.; Ren, Y.; Zhang, X.; Zheng, J.; Zhang, C.; Yang, C.; Mei, X.; et al. Whole transcriptome sequencing reveals that non-coding RNAs are related to embryo morphogenesis and development in rabbits. Genomics 2019, 112, 2203–2212. [Google Scholar] [CrossRef]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nature 2012, 14, 659–665. [Google Scholar] [CrossRef]
- Suvorova, I.I.; Grigorash, B.B.; Chuykin, I.A.; Pospelova, T.V.; Pospelov, V.A. G1 checkpoint is compromised in mouse ESCs due to functional uncoupling of p53-p21Waf1 signaling. Cell Cycle 2016, 15, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhu, Z.; Yu, F.; Huang, J.; Jia, R.; Pan, J. Effect of shRNA-mediated Xist knockdown on the quality of porcine parthenogenetic embryos. Dev. Dyn. 2019, 248, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Kirioukhova, O.; Shah, J.N.; Larsen, D.S.; Tayyab, M.; Mueller, N.E.; Govind, G.; Baroux, C.; Federer, M.; Gheyselinck, J.; Barrell, P.J.; et al. Aberrant imprinting may underlie evolution of parthenogenesis. Sci. Rep. 2018, 8, 10626. [Google Scholar] [CrossRef]
- Monteagudo-Sánchez, A.; Mora, J.R.H.; Simon, C.; Burton, A.; Tenorio, J.; Lapunzina, P.; Clark, S.; Esteller, M.; Kelsey, G.; López-Siguero, J.P.; et al. The role of ZFP57 and additional KRAB-zinc finger proteins in the maintenance of human imprinted methylation and multi-locus imprinting disturbances. Nucleic Acids Res. 2020, 48, 11394–11407. [Google Scholar] [CrossRef]
- Jiang, Z.; Dong, H.; Zheng, X.; Marjani, S.L.; Donovan, D.M.; Chen, J.; Tian, X. mRNA Levels of Imprinted Genes in Bovine In Vivo Oocytes, Embryos and Cross Species Comparisons with Humans, Mice and Pigs. Sci. Rep. 2015, 5, 17898. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, X.; Song, Y.; Lv, Q.; Lai, L.; Li, Z. Disruption of imprinted gene expression and DNA methylation status in porcine parthenogenetic fetuses and placentas. Gene 2014, 547, 351–358. [Google Scholar] [CrossRef]
- Nikaido, I.; Saito, C.; Mizuno, Y.; Meguro, M.; Bono, H.; Kadomura, M.; Kono, T.; Morris, G.A.; Lyons, P.A.; Oshimura, M.; et al. Discovery of Imprinted Transcripts in the Mouse Transcriptome Using Large-Scale Expression Profiling. Genome Res. 2003, 13, 1402–1409. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.; Li, J.; Zhang, X.; Chen, B.; Chi, D.; Zeng, Y.; Niu, Y.-J.; Wang, C.; Cheng, W.; Wu, W.; et al. Dynamic Reorganization of Nucleosome Positioning in Somatic Cells after Transfer into Porcine Enucleated Oocytes. Stem Cell Rep. 2017, 9, 642–653. [Google Scholar] [CrossRef] [Green Version]
- Teplitz, G.M.; Shi, M.; Sirard, M.A.; Lombardo, D.M. Coculture of porcine luteal cells during in vitro porcine oocyte maturation affects blastocyst gene expression and developmental potential. Theriogenology 2021, 166, 124–134. [Google Scholar] [CrossRef]
- Wang, T.; Gao, Y.-Y.; Chen, L.; Nie, Z.-W.; Cheng, W.; Liu, X.; Schatten, H.; Zhang, X.; Miao, Y.-L. Melatonin prevents postovulatory oocyte aging and promotes subsequent embryonic development in the pig. Aging 2017, 9, 1552–1564. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Liao, J.; Suen, A.H.C.; Lee, A.W.T.; Chung, H.S.; Tang, N.L.S.; Chow, K.L.; Cao, Q.; Yip, K.; Leung, T.Y.; et al. Comparative analysis of single-cell parallel sequencing approaches in oocyte application. Int. J. Biochem. Cell Biol. 2019, 107, 1–5. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Contreras-López, O.; Moyano, T.C.; Soto, D.C.; Gutiérrez, R.A. Step-by-Step Construction of Gene Co-expression Networks from High-Throughput Arabidopsis RNA Sequencing Data. Methods Mol. Biol. 2018, 1761, 275–301. [Google Scholar] [CrossRef]
- Trapnell, C.; A Williams, B.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Shi, G.; Chen, G.; Li, J.; Li, M.; Zou, C.; Fang, C.; Li, C. Transcriptome Analysis Suggests the Roles of Long Intergenic Non-coding RNAs in the Growth Performance of Weaned Piglets. Front. Genet. 2019, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Chen, L.; Chen, G.; Zou, C.; Li, J.; Li, M.; Fang, C.; Li, C. Identification and Functional Prediction of Long Intergenic Non-coding RNAs Related to Subcutaneous Adipose Development in Pigs. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhou, X.; Li, H.; Chen, Q.; Chen, G. Identification of the key genes and long non-coding RNAs in ankylosing spondylitis using RNA sequencing. Int. J. Mol. Med. 2018, 43, 1179–1192. [Google Scholar] [CrossRef]
- Kumar, H.; Srikanth, K.; Park, W.; Lee, S.-H.; Choi, B.-H.; Kim, H.; Kim, Y.M.; Cho, E.-S.; Kim, J.H.; Lee, J.H.; et al. Transcriptome analysis to identify long non coding RNA (lncRNA) and characterize their functional role in back fat tissue of pig. Gene 2019, 703, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Yang, D.; Liu, L.; Liu, Z.; Wang, J.; He, D.; Wu, H.; Wang, J.; Ma, Z. Genome-wide identification and characterization of long non-coding RNAs in MDCK cell lines with high and low tumorigenicities. Genomics 2019, 112, 1077–1086. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kuchipudi, S.V.; Tellabati, M.; Nelli, R.K.; White, G.A.; Perez, B.B.; Sebastian, S.; Slomka, M.J.; Brookes, S.M.; Brown, I.H.; Dunham, S.P.; et al. 18S rRNA is a reliable normalisation gene for real time PCR based on influenza virus infected cells. Virol. J. 2012, 9, 230. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Zheng, W.; Jin, C.; Zhan, Q.; Bai, L. Novel phenylpyrimidine derivatives containing a hydrazone moiety protect rice seedlings from injury by metolachlor. Bioorg. Chem. 2021, 108, 104645. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J.Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Liu, J. Transcriptome Analysis of Acid-Responsive Genes and Pathways Involved in Polyamine Regulation in Iron Walnut. Genes 2019, 10, 605. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Kim, D.G.; Park, M.C.; Um, J.Y.; Han, J.M.; Park, S.G.; Choi, E.C.; Kim, S. AIMP2 promotes TNFalpha-dependent apoptosis via ubiquitin-mediated degradation of TRAF2. J. Cell Sci. 2009, 122, 2710–2715. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.A.; Kura, N.; Sussman, C.; Woods, D.C. Mitochondrial membrane depolarization enhances TRAIL-induced cell death in adult human granulosa tumor cells, KGN, through inhibition of BIRC5. J. Ovarian Res. 2018, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wu, Y.; Li, Y.; Zheng, J.; Tang, J. A novel RING finger E3 ligase RNF186 regulate ER stress-mediated apoptosis through interaction with BNip1. Cell. Signal. 2013, 25, 2320–2333. [Google Scholar] [CrossRef]
- Luo, D.; Caniggia, I.; Post, M. Hypoxia-inducible regulation of placental BOK expression. Biochem. J. 2014, 461, 391–402. [Google Scholar] [CrossRef]
- Zou, C.; Li, L.; Cheng, X.; Li, C.; Fu, Y.; Fang, C.; Li, C. Identification and Functional Analysis of Long Intergenic Non-coding RNAs Underlying Intramuscular Fat Content in Pigs. Front. Genet. 2018, 9, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casero, D.; Sandoval, S.; Seet, C.S.; Scholes, J.; Zhu, Y.; Ha, V.L.; Luong, A.; Parekh, C.; Crooks, G.M. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat. Immunol. 2015, 16, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Ahn, R.; Gupta, R.; Lai, K.; Chopra, N.; Arron, S.T.; Liao, W. Network analysis of psoriasis reveals biological pathways and roles for coding and long non-coding RNAs. BMC Genom. 2016, 17, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.-G.; Liang, B.; Liu, J.-G.; Jin, S.-H.; He, S.-S.; Frey, B.; Gu, N.; Fietkau, R.; Hecht, M.; Ma, H.; et al. Identification of 15 lncRNAs Signature for Predicting Survival Benefit of Advanced Melanoma Patients Treated with Anti-PD-1 Monotherapy. Cells 2021, 10, 977. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xia, X.; Hu, Y.; Fang, X.; Orsulic, S. Identification of Infertility-Associated Topologically Important Genes Using Weighted Co-expression Network Analysis. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, H.; Wang, Q.; Guo, R.; Wei, L.; Song, H.; Kuang, W.; Liao, J.; Huang, Y.; Wang, Z. Genome-wide identification and characterization of long non-coding RNAs involved in flag leaf senescence of rice. Plant. Mol. Biol. 2021, 105, 655–684. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, X.; Tong, X.; Tang, C.; Li, J. Ppp2ca knockout in mice spermatogenesis. Reproduction 2015, 149, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Veil, M.; Schaechtle, M.A.; Gao, M.; Kirner, V.; Buryanova, L.; Grethen, R.; Onichtchouk, D. Maternal Nanog is required for zebrafish embryo architecture and for cell viability during gastrulation. Development 2018, 145, dev155366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, A.; Falace, A.; Wagner, M.; Gal, M.; Mei, D.; Conti, V.; Pisano, T.; Aprile, D.; Cerullo, M.S.; De Fusco, A.; et al. Biallelic DMXL2 mutations impair autophagy and cause Ohtahara syndrome with progressive course. Brain 2019, 142, 3876–3891. [Google Scholar] [CrossRef]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef]
- Zhou, Q.-Y.; Huang, J.-N.; Xiong, Y.-Z.; Zhao, S.-H. Imprinting analyses of the porcine GATM and PEG10 genes in placentas on days 75 and 90 of gestation. Genes Genet. Syst. 2007, 82, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallenberger, M.; Meinel, D.M.; Kroeber, M.; Wegner, M.; Milkereit, P.; Bösl, M.R.; Tamm, E.R. Lack of WDR36 leads to preimplantation embryonic lethality in mice and delays the formation of small subunit ribosomal RNA in human cells in vitro. Hum. Mol. Genet. 2010, 20, 422–435. [Google Scholar] [CrossRef] [Green Version]
- Humble, M.M.; Young, M.J.; Foley, J.F.; Pandiri, A.R.; Travlos, G.S.; Copeland, W.C. Polg2 is essential for mammalian embryogenesis and is required for mtDNA maintenance. Hum. Mol. Genet. 2012, 22, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinsson-Ahlzén, H.-S.; Liberal, V.; Grünenfelder, B.; Chaves, S.; Spruck, C.H.; Reed, S.I. Cyclin-Dependent Kinase-Associated Proteins Cks1 and Cks2 Are Essential during Early Embryogenesis and for Cell Cycle Progression in Somatic Cells. Mol. Cell. Biol. 2008, 28, 5698–5709. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhu, Y.; Li, Z.; Bu, Q.; Sun, T.; Wang, H.; Sun, H.; Cao, X. Up-regulation of SPC25 promotes breast cancer. Aging 2019, 11, 5689–5704. [Google Scholar] [CrossRef] [PubMed]
- Legrand, J.M.D.; Chan, A.-L.; La, H.M.; Rossello, F.J.; Änkö, M.-L.; Fuller-Pace, F.V.; Hobbs, R.M. DDX5 plays essential transcriptional and post-transcriptional roles in the maintenance and function of spermatogonia. Nat. Commun. 2019, 10, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishishita, S.; Matsuda, Y.; Kitada, K. Genetic evidence suggests that Spata22 is required for the maintenance of Rad51 foci in mammalian meiosis. Sci. Rep. 2014, 4, srep06148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Ortega, M.A.; Ko, M.; Marh, J.; Ward, W.S. ORC4 Surrounds Extruded Chromatin in Female Meiosis. J. Cell. Biochem. 2014, 116, 778–786. [Google Scholar] [CrossRef] [Green Version]
- Wojtanowicz-Markiewicz, K.; Kocherova, I.; Jeseta, M.; Piotrowska-Kempisty, H.; Brüssow, K.P.; Skowroński, M.T.; Bruska, M.; Bukowska, D.; Nowicki, M.; Kempisty, B.; et al. Expression of PTX3, HAS2 AND TNFAIP6 genes in relation to real-time proliferation of porcine endometrial luminal epithelial cells in primary cultivation model. J. Boil. Regul. Homeost. Agents 2019, 33, 675–685. [Google Scholar]
- Towler, B.P.; Jones, C.I.; Harper, K.L.; Waldron, J.A.; Newbury, S.F. A novel role for the 3′-5′ exoribonuclease Dis3L2 in controlling cell proliferation and tissue growth. RNA Biol. 2016, 13, 1286–1299. [Google Scholar] [CrossRef]
- Luo, T.; Xu, Y.; Hoffman, T.L.; Zhang, T.; Schilling, T.; Sargent, T.D. Inca: A novel p21-activated kinase-associated protein required for cranial neural crest development. Development 2007, 134, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Cang, Y.; Zhang, J.; Nicholas, S.A.; Bastien, J.; Li, B.; Zhou, P.; Goff, S.P. Deletion of DDB1 in Mouse Brain and Lens Leads to p53-Dependent Elimination of Proliferating Cells. Cell 2006, 127, 929–940. [Google Scholar] [CrossRef] [Green Version]
- Ishimura, A.; Minehata, K.-I.; Terashima, M.; Kondoh, G.; Hara, T.; Suzuki, T. Jmjd5, an H3K36me2 histone demethylase, modulates embryonic cell proliferation through the regulation of Cdkn1a expression. Development 2012, 139, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Kono, T. Genomic imprinting is a barrier to parthenogenesis in mammals. Cytogenet. Genome Res. 2006, 113, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.R.; Tsai, S.; Hardison, N.; Motsinger-Reif, A.A.; Freking, B.A.; Nonneman, D.; Rohrer, G.; Piedrahita, J.A. Characterization of Conserved and Nonconserved Imprinted Genes in Swine1. Biol. Reprod. 2009, 81, 906–920. [Google Scholar] [CrossRef] [Green Version]
- Warzych, E.; Peippo, J.; Szydlowski, M.; Lechniak, D.M. Supplements to in vitro maturation media affect the production of bovine blastocysts and their apoptotic index but not the proportions of matured and apoptotic oocytes. Anim. Reprod. Sci. 2007, 97, 334–343. [Google Scholar] [CrossRef]
- Gomez, E.; Gutierrez-Adan, A.; Díez, C.; Bermejo-Álvarez, P.; Muñoz, M.; Rodriguez, A.; Otero, J.; Alvarez-Viejo, M.; Martín, D.; Carrocera, S.; et al. Biological differences between in vitro produced bovine embryos and parthenotes. Reproduction 2009, 137, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, B.J.; Jang, H.S.; Song, H.; Park, C.; Hong, K.; Lee, J.W.; Do, J.T. Generation of Mouse Parthenogenetic Epiblast Stem Cells and Their Imprinting Patterns. Int. J. Mol. Sci. 2019, 20, 5428. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, N.; Wang, D.; Han, X.; Lv, Q.; Ouyang, H.; Li, Z. Isoform-specific imprinting of the MEST gene in porcine parthenogenetic fetuses. Gene 2015, 558, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.; Nakamura, K.; Inoue, K.; Naruse, M.; Usami, T.; Wakisaka-Saito, N.; Hino, T.; Suzuki-Migishima, R.; Ogonuki, N.; Miki, H.; et al. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat. Genet. 2005, 38, 101–106. [Google Scholar] [CrossRef]
- Adnani, L.; Langevin, L.M.; Gautier, E.; Dixit, R.; Parsons, K.; Li, S.; Kaushik, G.; Wilkinson, G.; Wilson, R.; Childs, S.; et al. Zac1 Regulates the Differentiation and Migration of Neocortical Neurons via Pac1. J. Neurosci. 2015, 35, 13430–13447. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Embryo Source | Total Embryos (0 h) | Cleaved Embryos (48 h) | Blastocysts (6–7 d) |
|---|---|---|---|
| IVF | 125 | 108 (86.38%) | 33 (30.56% a –26.40% b) |
| 305 | 183 (60.00%) | 49 (26.77% a –16.07% b) | |
| 129 | 86 (66.67%) | 19 (22.09% a –14.73% b) | |
| PA | 55 | 46 (83.64%) | 15 (32.60% a –27.27% b) |
| 47 | 43 (91.49%) | 11 (25.58% a –23.40% b) | |
| 49 | 34 (69.39%) | 13 (38.24% a –26.53% b) |
| Sample Name | Raw Reads | Clean Reads | Clean Bases | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) | Uniquely Mapping Rate | Overall Alignment Rate |
|---|---|---|---|---|---|---|---|---|---|
| IVF_Z_1 | 137468078 | 104586902 | 13.01 G | 0.03 | 96.73 | 92.23 | 44.97 | 65.65% | 92.07% |
| IVF_Z_2 | 142771986 | 100439624 | 12.43 G | 0.02 | 97.53 | 94.89 | 44.98 | 74.25% | 91.80% |
| IVF_Z_3 | 135512064 | 102006570 | 12.63 G | 0.02 | 96.36 | 92.48 | 46.97 | 73.66% | 94.41% |
| IVF_M_1 | 156959598 | 98837458 | 12.36 G | 0.04 | 98.78 | 96.27 | 45.57 | 74.38% | 95.23% |
| IVF_M_2 | 169026320 | 105937556 | 13.09 G | 0.03 | 97.59 | 94.78 | 46.02 | 65.17% | 92.75% |
| IVF_M_3 | 156859688 | 100002906 | 12.37 G | 0.03 | 97.7 | 95.13 | 47.08 | 66.95% | 92.47% |
| IVF_B_1 | 150759132 | 99269248 | 12.33 G | 0.02 | 97.74 | 94.3 | 48.21 | 70.28% | 94.86% |
| IVF_B_2 | 181545464 | 115480292 | 14.26 G | 0.02 | 95.78 | 92.17 | 39.67 | 63.31% | 92.36% |
| IVF_B_3 | 146705060 | 100166918 | 12.38 G | 0.02 | 98.09 | 95.89 | 49.42 | 71.38% | 93.21% |
| MII_1 | 139913758 | 104812570 | 13.01 G | 0.03 | 95.55 | 90.93 | 45.49 | 76.37% | 95.77% |
| MII_2 | 165517814 | 112417876 | 14 G | 0.02 | 98.04 | 95.7 | 45.71 | 47.38% | 82.04% |
| MII_3 | 176029002 | 129015192 | 16 G | 0.02 | 97.92 | 95.13 | 45.12 | 69.03% | 93.92% |
| PA_Z_1 | 136106014 | 100661316 | 12.5 G | 0.03 | 95.95 | 91.54 | 46 | 69.40% | 92.33% |
| PA_Z_2 | 150617128 | 109910972 | 13.67 G | 0.02 | 97.88 | 95.13 | 45.91 | 74.20% | 94.61% |
| PA_Z_3 | 146127012 | 99562818 | 12.38 G | 0.02 | 97.69 | 94.81 | 46.23 | 75.18% | 94.60% |
| PA_M_1 | 223196572 | 100931396 | 12.42 G | 0.04 | 97.52 | 94.85 | 44.97 | 65.31% | 91.52% |
| PA_M_2 | 188725788 | 101995272 | 12.54 G | 0.03 | 96.72 | 93.15 | 42.28 | 56.11% | 89.39% |
| PA_M_3 | 181377016 | 110631938 | 13.57 G | 0.03 | 95.48 | 91.64 | 43.6 | 47.55% | 82.89% |
| PA_B_1 | 197029468 | 123993046 | 15.39 G | 0.02 | 98.76 | 97.11 | 47.02 | 76.96% | 95.96% |
| PA_B_2 | 219332164 | 144578468 | 17.82 G | 0.02 | 95.57 | 91.51 | 42.61 | 51.02% | 85.29% |
| PA_B_3 | 182772226 | 138010990 | 17.07 G | 0.02 | 95.79 | 91.62 | 44.59 | 55.11% | 87.73% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zou, C.; Li, M.; Fang, C.; Li, K.; Liu, Z.; Li, C. Transcriptome Analysis of In Vitro Fertilization and Parthenogenesis Activation during Early Embryonic Development in Pigs. Genes 2021, 12, 1461. https://doi.org/10.3390/genes12101461
Li X, Zou C, Li M, Fang C, Li K, Liu Z, Li C. Transcriptome Analysis of In Vitro Fertilization and Parthenogenesis Activation during Early Embryonic Development in Pigs. Genes. 2021; 12(10):1461. https://doi.org/10.3390/genes12101461
Chicago/Turabian StyleLi, Xin, Cheng Zou, Mengxun Li, Chengchi Fang, Kui Li, Zhiguo Liu, and Changchun Li. 2021. "Transcriptome Analysis of In Vitro Fertilization and Parthenogenesis Activation during Early Embryonic Development in Pigs" Genes 12, no. 10: 1461. https://doi.org/10.3390/genes12101461
APA StyleLi, X., Zou, C., Li, M., Fang, C., Li, K., Liu, Z., & Li, C. (2021). Transcriptome Analysis of In Vitro Fertilization and Parthenogenesis Activation during Early Embryonic Development in Pigs. Genes, 12(10), 1461. https://doi.org/10.3390/genes12101461