
Differential Diagnosis between Marfan Syndrome and Loeys–Dietz Syndrome Type 4: A Novel Chromosomal Deletion Covering TGFB2


 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
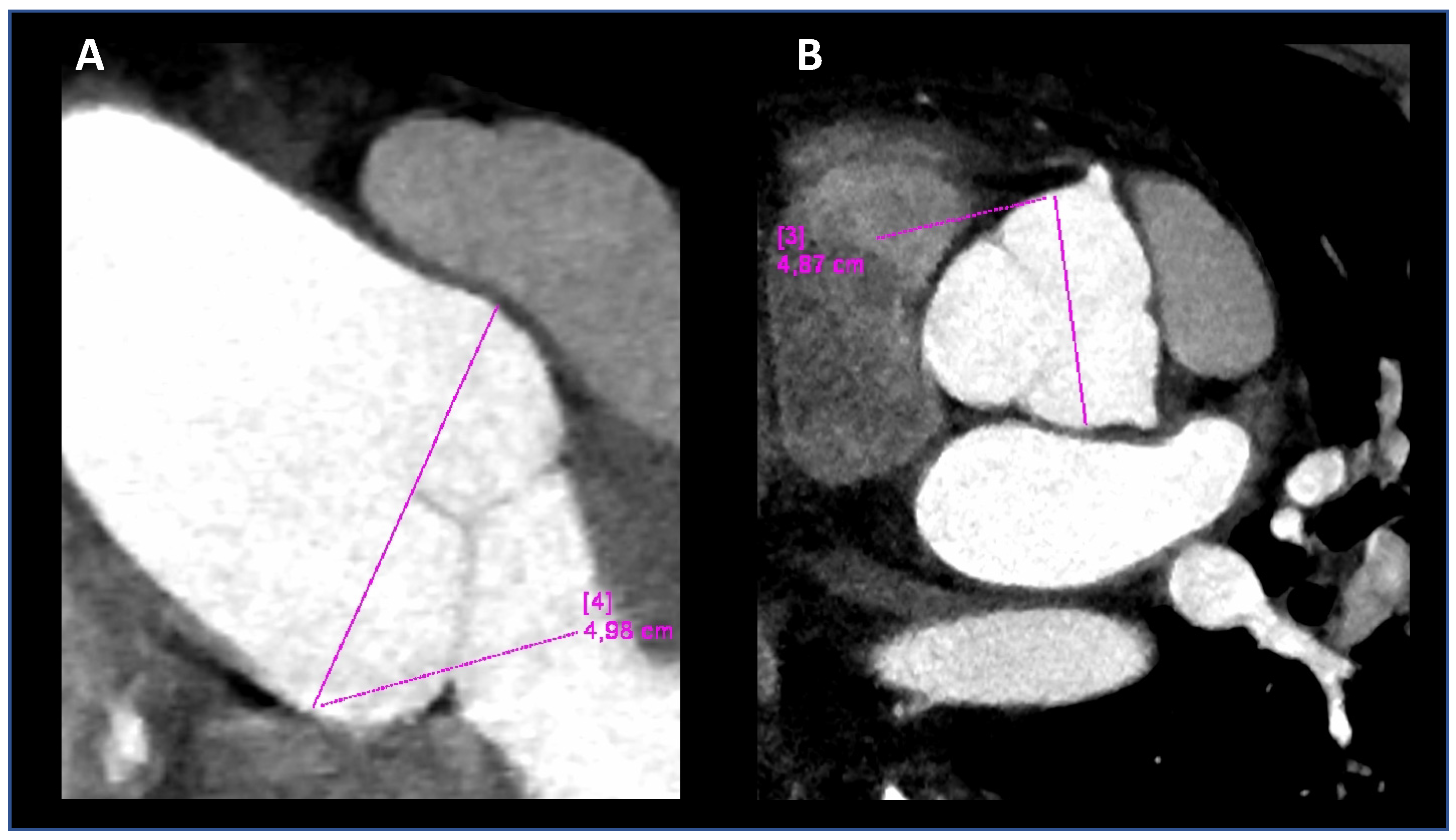
2.2. Imaging Analysis
2.3. Genomic DNA Preparation
2.4. Next Generation Sequencing (NGS) Analysis
2.5. Alignment and Variants Calling
2.6. Array-CGH Analyses
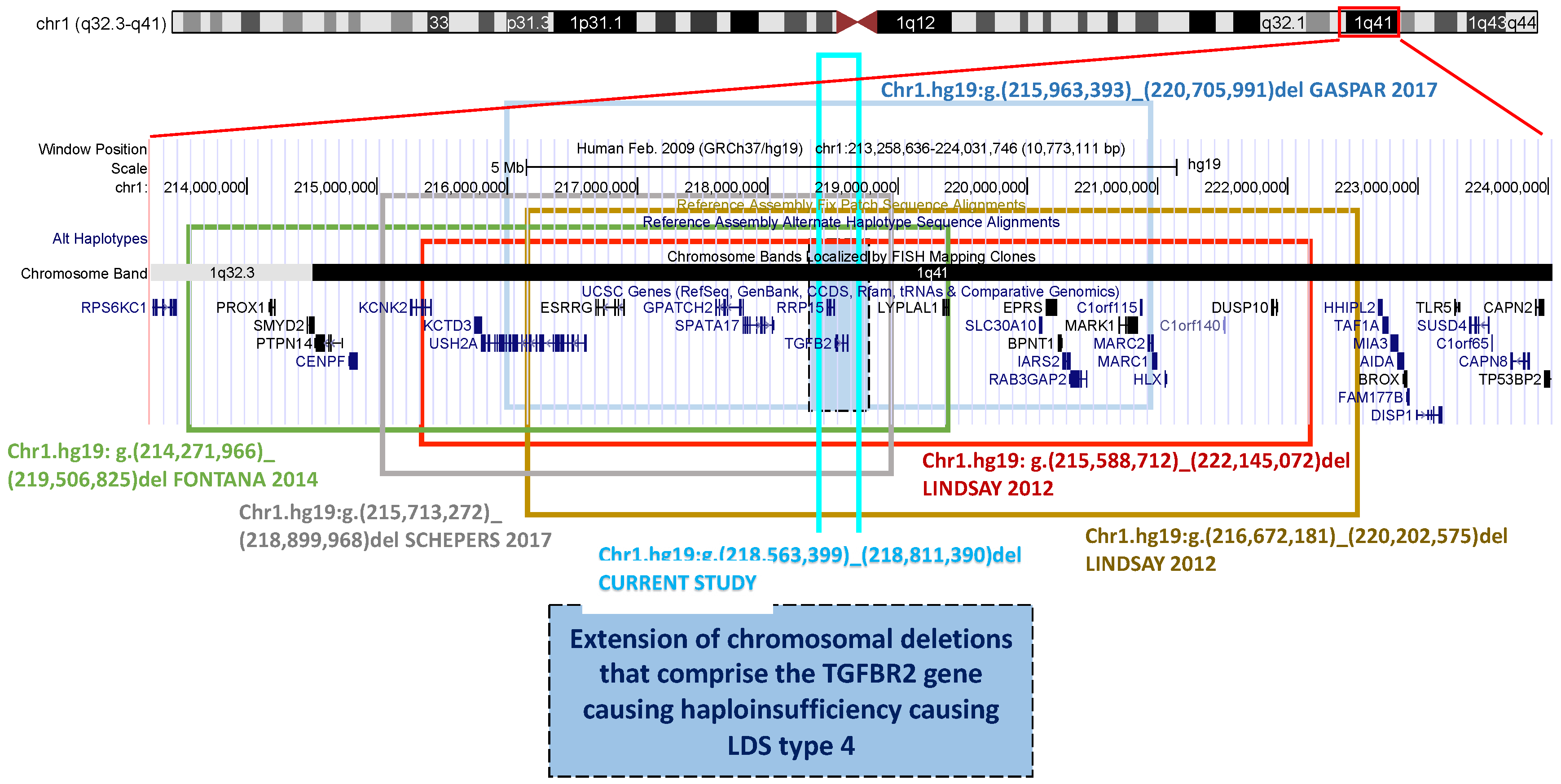
3. Results
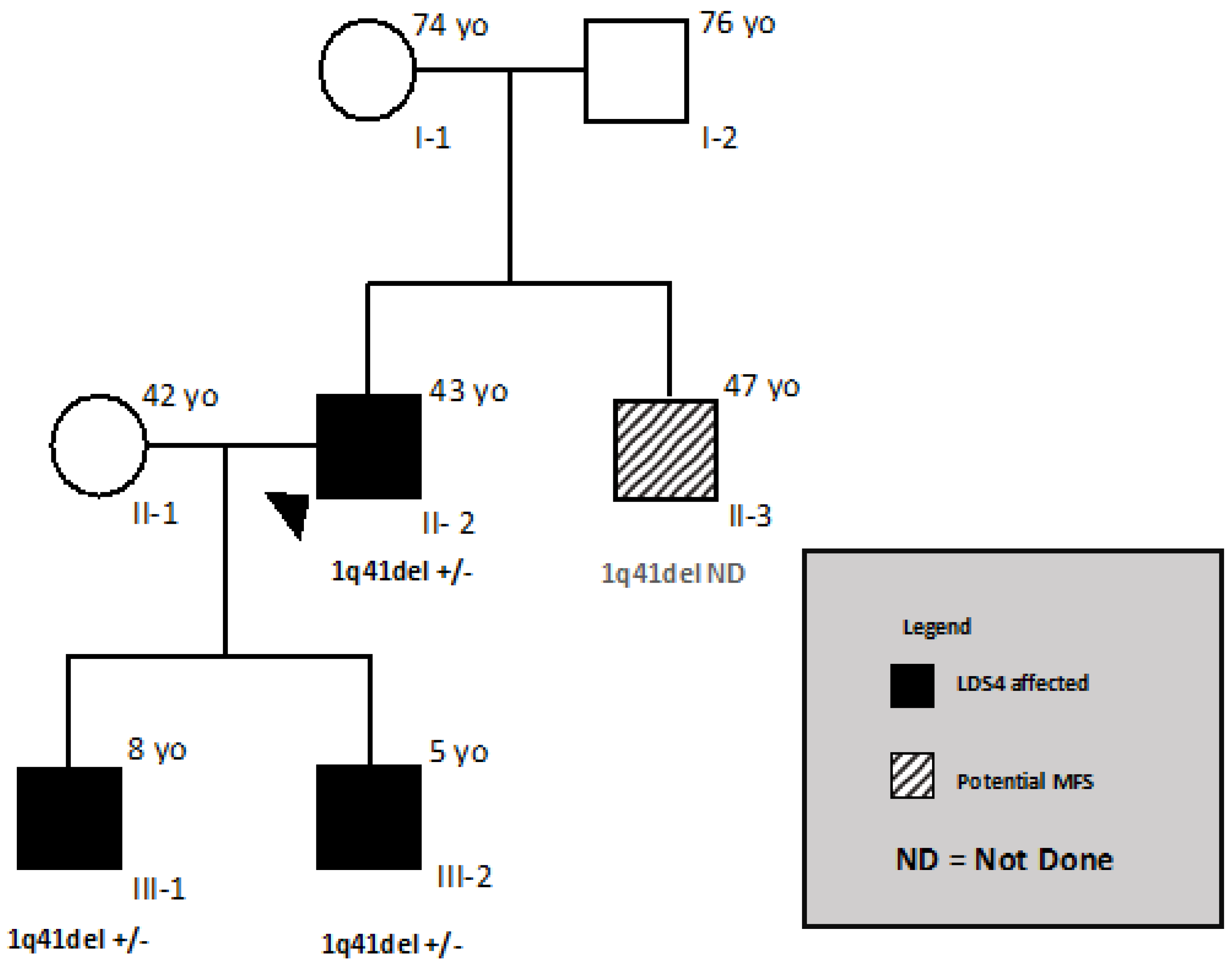
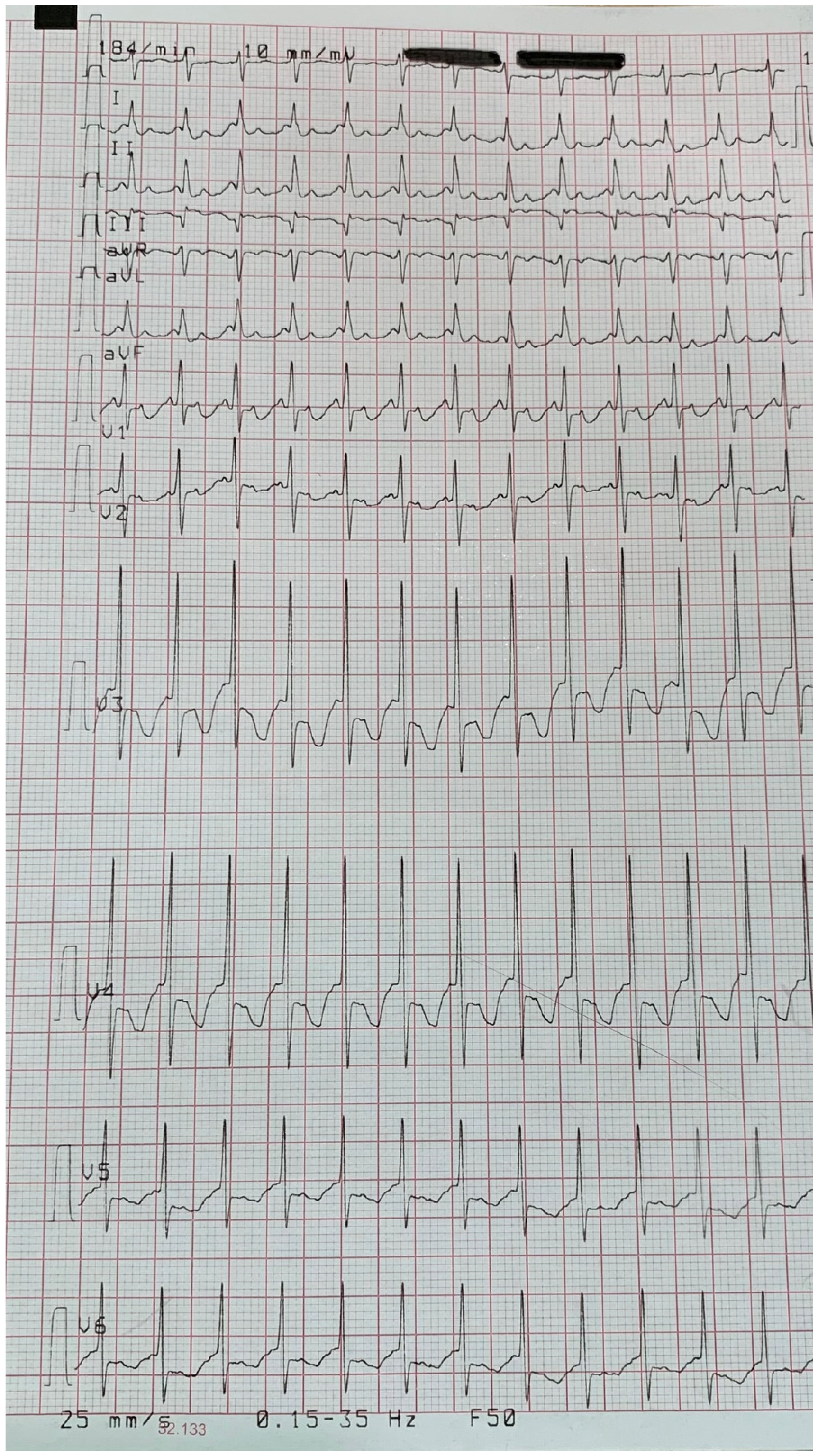
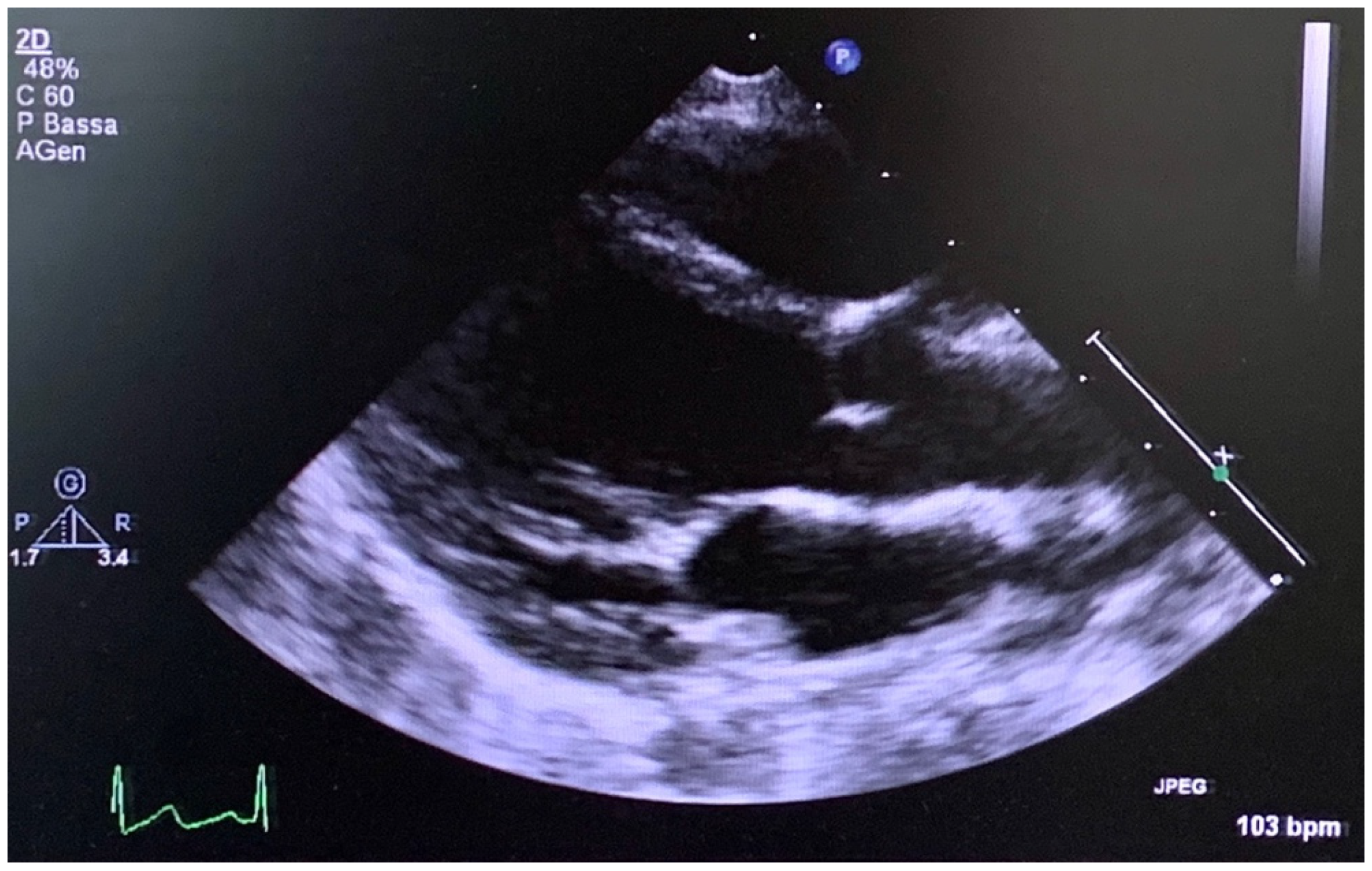
3.1. Patient and His Family
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Gene 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.; de Graaf, B.; Verhagen, J.; Hoedemaekrs, Y.; Willemsen, R.; Severijnen, L.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and acute aortic dissections associated with mild systemic features of the Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.A.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.T.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeys, B.L.; Dietz, H.C. Loeys-Dietz Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2008; pp. 1993–2021. [Google Scholar]
- Fontana, P.; Genesio, R.; Casertano, A.; Cappuccio, G.; Mormile, A.; Nitsch, L.; Iolascon, A.; Andria, G.; Melis, D. Loeys-Dietz syndrome type 4, caused by chromothripsis, involving the TGFB2 gene. Gene 2014, 538, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.; Lutz, B.; Reicherter, K.; Lühl, S.; Taurman, R.; Gabriel, H.; Brenner, R.E.; Borck, G. 4.7 Mb deletion encompassing TGFB2 associated with features of Loeys-Dietz syndrome and osteoporosis in adulthood. Am. J. Med. Genet. A 2017, 173, 2289–2292. [Google Scholar] [CrossRef] [PubMed]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, M.J.; Devereux, R.B.; Kramer-Fox, R.; O’Loughlin, J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am. J. Cardiol. 1989, 64, 507–512. [Google Scholar] [CrossRef]
- Gautier, M.; Detaint, D.; Fermanian, C.; Aegerter, P.; Delorme, G.; Arnoult, F.; Milleron, O.; Raoux, F.; Stheneur, C.; Boileau, C.; et al. Nomograms for aortic root diameters in children using two-dimensional echocardiography. Am. J. Cardiol. 2010, 105, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Giusti, B.; Sticchi, E.; De Cario, R.; Magi, A.; Nistri, S.; Pepe, G. Genetic Bases of Bicuspid Aortic Valve: The Contribution of Traditional and High-Throughput Sequencing Approaches on Research and Diagnosis. Front. Physiol. 2017, 8, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giunti, L.; Pantaleo, M.; Sardi, I.; Provenzano, A.; Magi, A.; Cardellicchio, S.; Castiglione, F.; Tattini, L.; Novara, F.; Buccoliero, A.M.; et al. Genome-wide copy number analysis in pediatric glioblastoma multiforme. Am. J. Cancer Res. 2014, 4, 293–303. [Google Scholar] [PubMed]
- Braverman, A.C.; Blinder, K.J.; Khanna, S.; Willing, M. Ectopia lentis in Loeys-Dietz syndrome type 4. Am. J. Med. Genet. A 2020, 182, 1957–1959. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Manifestations | MFS | LDS4 | Figure 1, This Report | |||
|---|---|---|---|---|---|---|
| Genes | FBN1 | TGFB2 | II-2 | II-3 | ||
| Systemic Features (1) Score =/> 7 | Score of SF | Systemic Features | Clinical Features | Clinical Features | Score of SF | |
| Facial Features: | 1 | 1 | ||||
| 1 | dolichocephaly | -- | + | |||
| If present 3/5 features | downslanting palpebral fissures | + | -- | |||
| enophthalmos | + | + | ||||
| malar hypoplasia | + | + | ||||
| retrognatia | + | + | ||||
| Body segments | 1 | Reduced US/LS AND increased arm span (AS)/height (H) AND no severe scoliosis | 1 (US/LS0.54, AS/H = 1.06) | -- | ||
| + | + | |||||
| Pectus deformity | ||||||
| 2 | carinatum | ++ | ++ | 2 | -- | |
| 1 | Excavatum or chest asimmetry | -- | 1 | |||
| Rachis | 1 | >20 °C Scoliosis or thoracolumbar kyphosis | + | -- | ||
| Upper limb | 1 | Reduced elbow extension | -- | 1 | ||
| 3 | Wrist AND thumb sign | 3 | -- | |||
| 1 | (wrist OR thumb sign | |||||
| 2 | Protrusio acetabuli | N.A. | N.A. | |||
| Lower limb | 2 | Hindfoot deformity | -- | 2 | ||
| 1 | plain pes planus | 1 | 1 | |||
| 2 | Dural ectasia (DE) | + | + | -- | N.A. | |
| 2 | Pneumotorax (PNX) | -- | -- | |||
| 1 | Mitral Valve Prolapse (MVP, any type) | -- | -- | |||
| 1 | Myopia >3Ds | + | + | |||
| 1 | Skin striae | ++ | + | 1 | 1 | |
| CVS | Aortic root aneurysms (2) | ++ | ++ | + | -- | |
| TAD (2) | + | + | -- | -- | ||
| Other CVS | Ascending aorta aneurysm | -- | -- | |||
| Other aneurysms | + | + | + late onset | -- | ||
| Arterial tortuosity | - | + | + late onset | -- | ||
| BAV(bicuspid aortic valve) | + | ++ | -- | -- | ||
| (C) Eyes | Ectopia lentis (EL) (3) | +++ | -- | -- | -- | |
| Cleft palalate/bifid uvula | -- | + | -- | -- | ||
| Hypertelorysm | -- | + | -- | -- | ||
| Tall stature | +++ | ++ | + | + | ||
| Arachnodactyly | +++ | + | + hands and feet | + | ||
| Clubfoot | -- | ++ | -- | -- | ||
| Osteoarthritis | ++ | + | -- | -- | ||
| Hernia | + | + | + | -- | ||
| Hypermobility | + | -- | ||||
| GENES | FBN1 | - | -- | |||
| TGFB2arrayCGH | + | N.A | ||||
| TGFB2 NGS | - | N.A. | ||||
| Reference and PatientID | Lindsay 2012 II-1 | Lindsay 2012 II-2 | Fontana 2014 | Gaspar 2017 II-2 | Gaspar 2017 III-1 | This Report |
|---|---|---|---|---|---|---|
| Mutation | 1pq41 ch del | 1pq41 ch del | 1pq41 ch del | 1pq41 ch del | 1pq41 ch del | 1pq41 ch del |
| Deletion size | 6.5 Mb | 3.5 Mb | 5.2 Mb | 4.7 Mb | 4.7 Mb | 0.25 Mb |
| Number of deleted genes known to encode proteins | 20 | 9 | 15 | 18 | 18 | 2 |
| Sex | M | M | F | F | M | M |
| Age | 46 | 9 | 18 | 40 | 12 | 43 |
| Craniofacial | ||||||
| Eye | myopia | hyperopia | Severe myopia, strabismus, exotropia, ptosis, nystagmus progressive tapeto-retinal degeneration, blue sclera | - | - | myopia |
| Downslant palpebral fissures | - | + | + | + | + | + |
| Hypertelorism | + | - | + | - | - | - |
| High arched palate | + | + | + | - | - | + |
| Uvula | N.R. | N.R. | - | - | - | - |
| Retrognathia | + | + | + | N.R. | + | + |
| OTHER | torticollis | ptosis | Triangular face, low-set ears, thin lips, mild conductive hypoacusis, dental enamel hypoplasia | Dental enamel hypoplasia, abn, anteversal nares | ||
| Skeletal | ||||||
| Stature cm or percentile | 193 | 195 | 145.7 | 154 | 200 | |
| Armspan ratio | 1.02 | 0.96 | N.R. | N.R. | 1.04 | |
| Pectus Deformity | + | + | + | N.R. | + | + |
| Scoliosis | + | - | + Dorso-lumbar scoliosis | N.R. | N.R. | + |
| Arachnodactily | + | + | N.R. | + | - | + |
| Positive thumb/wrist | - | - | - | + | - | + |
| Club feet | + | + | - | - | N.R. | - |
| Pes planus | - | + | - | N.R. | N.R. | - |
| Joint Hypermobility > 5/9 | N.R. | N.R. | N.R. | + (7/9) | + (6/9) | + (8/9) |
| OTHER | Mild motor, language delay, optic canal hyperostosis, coxa vara, genu varu, coxa valga surgery, delta-storage pool pt disease | Osteoporosis bilateral femoral and neck, fractures of the pelvis and the ribs, muscle weakness, chronic pain | Muscular hypotonia, problems with motor coordination, dyslalia | |||
| Cardiovascular | ||||||
| Aortc root-z-score | 2.8 | 3.0 | 2.35 | N.R. | - | 5.0 before surgery |
| Ao | - | - | 31 normal size | Normal size | Normal size | Surgery at 38 years |
| Ao dissection/repair | typeB, age42 | - | - | - | - | |
| Aortic valve | TAV | TAV | TAV | TAV | TAV | TAV |
| Mitral valve | MVP | - | - Redundant cusps | - | - | - |
| Arterial aneurysm | - | - | - | - | - | Left common iliac, abdominal aorta, mild ectasia 14 × 16 (mm) |
| Arterial tortuosity | - | - | - | - | - | Arterial tortuosity iliacs |
| OTHER | Claw toes | Claw toes | ||||
| Skin | ||||||
| Striae | - | - | - | - | - | + |
| Hernia | + | - | - | - | + inguinal right surgery | + |
| Easy bruising | - | - | + | - | + | - |
| OTHER | Hematomas, nose bleeding | |||||
| dura | ND | ND | ND | ND | ND | Periradicular cysts |
| Other findings | cryptorchidism | Hypotonia, ataxia | Epileptic seizures | Excluded learning disability | hypothyroidism | Varicose veins, deep venous thrombosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nistri, S.; De Cario, R.; Sticchi, E.; Spaziani, G.; Della Monica, M.; Giglio, S.; Favilli, S.; Giusti, B.; Stefano, P.; Pepe, G. Differential Diagnosis between Marfan Syndrome and Loeys–Dietz Syndrome Type 4: A Novel Chromosomal Deletion Covering TGFB2. Genes 2021, 12, 1462. https://doi.org/10.3390/genes12101462
Nistri S, De Cario R, Sticchi E, Spaziani G, Della Monica M, Giglio S, Favilli S, Giusti B, Stefano P, Pepe G. Differential Diagnosis between Marfan Syndrome and Loeys–Dietz Syndrome Type 4: A Novel Chromosomal Deletion Covering TGFB2. Genes. 2021; 12(10):1462. https://doi.org/10.3390/genes12101462
Chicago/Turabian StyleNistri, Stefano, Rosina De Cario, Elena Sticchi, Gaia Spaziani, Matteo Della Monica, Sabrina Giglio, Silvia Favilli, Betti Giusti, Pierluigi Stefano, and Guglielmina Pepe. 2021. "Differential Diagnosis between Marfan Syndrome and Loeys–Dietz Syndrome Type 4: A Novel Chromosomal Deletion Covering TGFB2" Genes 12, no. 10: 1462. https://doi.org/10.3390/genes12101462
APA StyleNistri, S., De Cario, R., Sticchi, E., Spaziani, G., Della Monica, M., Giglio, S., Favilli, S., Giusti, B., Stefano, P., & Pepe, G. (2021). Differential Diagnosis between Marfan Syndrome and Loeys–Dietz Syndrome Type 4: A Novel Chromosomal Deletion Covering TGFB2. Genes, 12(10), 1462. https://doi.org/10.3390/genes12101462