
A Familial Form of Epidermolysis Bullosa Simplex Associated with a Pathogenic Variant in KRT5

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient and Family Members
2.2. Immunohistochemistry (IHC) and Immunofluorescence (IF)
2.3. Molecular Analysis
2.4. Statement of Ethics
3. Results

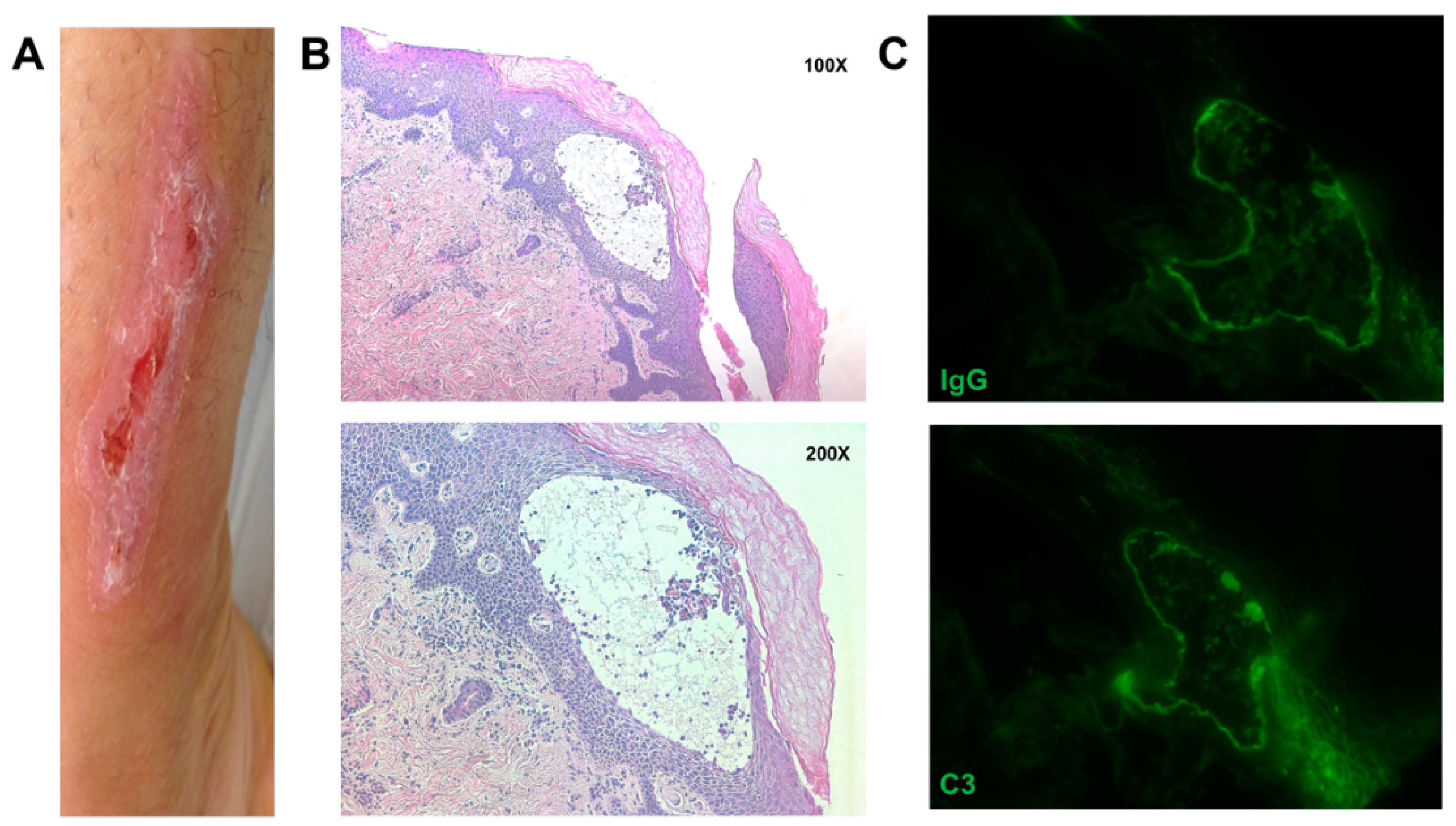{kind=link}
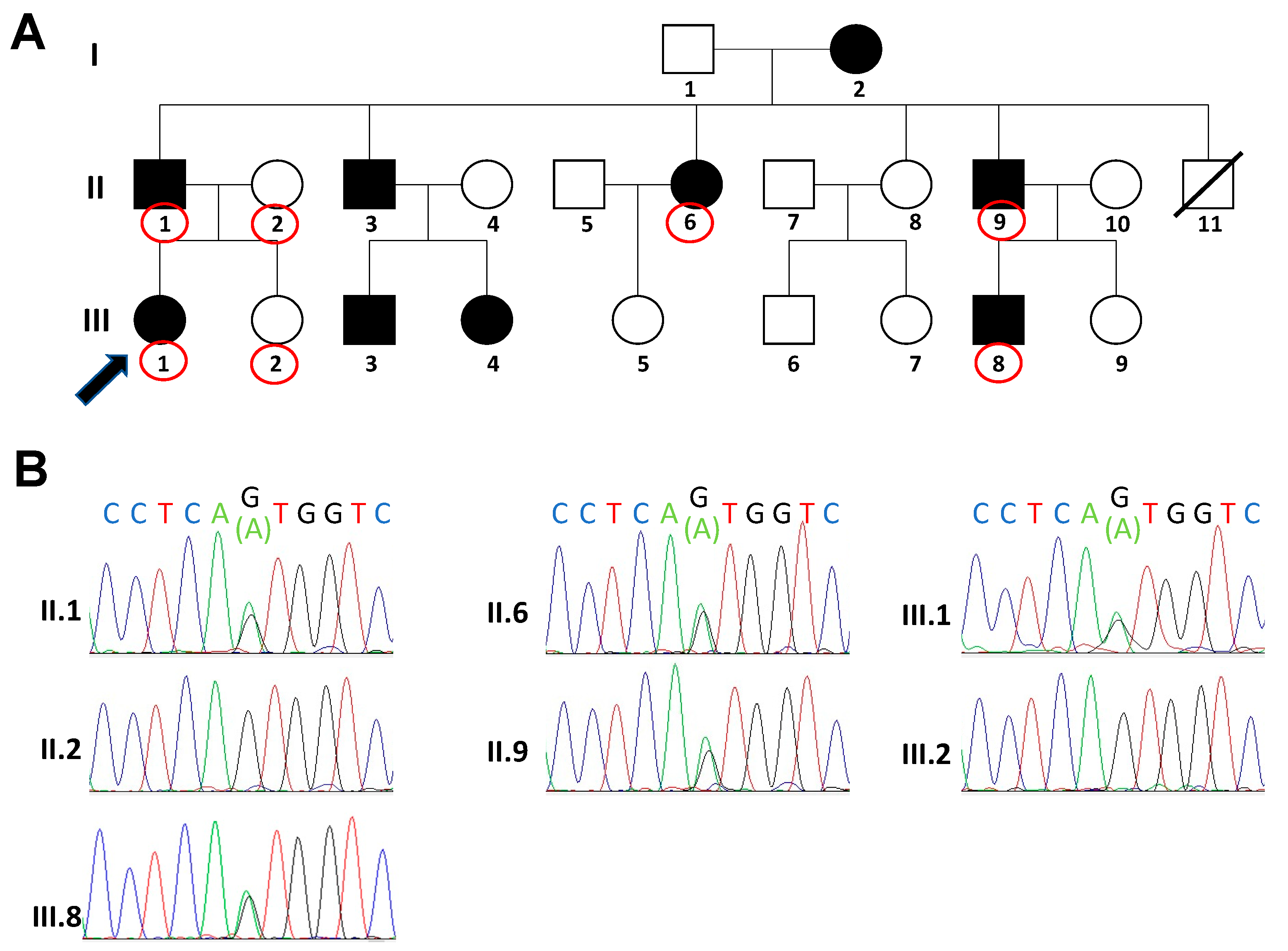{kind=link}
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bardhan, A.; Bruckner-Tuderman, L.; Chapple, I.L.; Fine, J.-D.; Harper, N.; Has, C.; Magin, T.M.; Marinkovich, M.P.; Marshall, J.F.; McGrath, J.A. Epidermolysis bullosa. Nat. Rev. Dis. Primers 2020, 6, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Intong, L.R.; Murrell, D.F. Inherited epidermolysis bullosa: New diagnostic criteria and classification. Clin. Dermatol. 2012, 30, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Pfendner, E.G.; Sadowski, S.G.; Uitto, J. Epidermolysis bullosa simplex: Recurrent and de novo mutations in the KRT5 and KRT14 genes, phenotype/genotype correlations, and implications for genetic counseling and prenatal diagnosis. J. Investig. Dermatol. 2005, 125, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Mariath, L.M.; Santin, J.T.; Schuler-Faccini, L.; Kiszewski, A.E. Inherited epidermolysis bullosa: Update on the clinical and genetic aspects☆,☆☆. An. Bras. De Dermatol. 2020, 95, 551–569. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.; Bodemer, C.; Bolling, M.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Kerns, M.L.; Fuchs, E. Epidermolysis bullosa simplex: A paradigm for disorders of tissue fragility. J. Clin. Investig. 2009, 119, 1784–1793. [Google Scholar] [CrossRef]
- Fuchs, E.; Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Hsu, S.M.; Lee, J.Y.Y.; Yang, M.H.; Chao, S.C. A novel mutation in the L12 domain of keratin 5 in the Köbner variant of epidermolysis bullosa simplex. Dermatol. Sin. 2005, 23, 32–35. [Google Scholar]
- Scali, E.; Mignogna, C.; Di Vito, A.; Presta, I.; Camastra, C.; Donato, G.; Bottoni, U. Inflammation and macrophage polarization in cutaneous melanoma: Histopathological and immunohistochemical study. Int. J. Immunopathol. Pharmacol. 2016, 29, 715–719. [Google Scholar] [CrossRef]
- Paduano, F.; Colao, E.; Loddo, S.; Orlando, V.; Trapasso, F.; Novelli, A.; Perrotti, N.; Iuliano, R. 7q35 microdeletion and 15q13. 3 and xp22. 33 microduplications in a patient with severe myoclonic epilepsy, microcephaly, dysmorphisms, severe psychomotor delay and intellectual disability. Genes 2020, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Paduano, F.; Fabiani, F.; Colao, E.; Trapasso, F.; Perrotti, N.; Barbieri, V.; Baudi, F.; Iuliano, R. Case Report: Identification of a Novel Pathogenic Germline TP53 Variant in a Family With Li–Fraumeni Syndrome. Front. Genet. 2021, 1541. [Google Scholar] [CrossRef]
- Schuilenga-Hut, P.H.; Vlies, P.v.; Jonkman, M.F.; Waanders, E.; Buys, C.H.; Scheffer, H. Mutation analysis of the entire keratin 5 and 14 genes in patients with epidermolysis bullosa simplex and identification of novel mutations. Hum. Mutat. 2003, 21, 447. [Google Scholar] [CrossRef]
- Patel, P.M.; Jones, V.A.; Behnam, C.T.; Zenzo, G.D.; Amber, K.T. A Review of Acquired Autoimmune Blistering Diseases in Inherited Epidermolysis Bullosa: Implications for the Future of Gene Therapy. Antibodies 2021, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Szeverenyi, I.; Cassidy, A.J.; Chung, C.W.; Lee, B.T.; Common, J.E.; Ogg, S.C.; Chen, H.; Sim, S.Y.; Goh, W.L.; Ng, K.W. The Human Intermediate Filament Database: Comprehensive information on a gene family involved in many human diseases. Hum. Mutat. 2008, 29, 351–360. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Galligan, P.; Listwan, P.; Siller, G.M.; Rothnagel, J.A. A novel mutation in the L12 domain of keratin 5 in the Köbner variant of epidermolysis bullosa simplex. J. Investig. Dermatol. 1998, 111, 524–527. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, E.N.; Harris, A.G.; Bingham, L.J.; Yan, W.; Su, J.C.; Murrell, D.F. A review of 52 pedigrees with epidermolysis bullosa simplex identifying ten novel mutations in KRT5 and KRT14 in Australia. Acta Derm.-Venereol. 2017, 97, 1114–1119. [Google Scholar] [CrossRef]
- Bolling, M.; Lemmink, H.; Jansen, G.; Jonkman, M. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br. J. Dermatol. 2011, 164, 637–644. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Lee, C.-H. Defining keratin protein function in skin epithelia: Epidermolysis bullosa simplex and its aftermath. J. Investig. Dermatol. 2012, 132, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Wertheim-Tysarowska, K.; Ołdak, M.; Giza, A.; Kutkowska-Kaźmierczak, A.; Sota, J.; Przybylska, D.; Woźniak, K.; Śniegórska, D.; Niepokój, K.; Sobczyńska-Tomaszewska, A. Novel sporadic and recurrent mutations in KRT5 and KRT14 genes in Polish epidermolysis bullosa simplex patients: Further insights into epidemiology and genotype–phenotype correlation. J. Appl. Genet. 2016, 57, 175–181. [Google Scholar] [CrossRef]
- Rugg, E.L.; Horn, H.M.; Smith, F.J.; Wilson, N.J.; Hill, A.J.; Magee, G.J.; Shemanko, C.S.; Baty, D.U.; Tidman, M.J.; Lane, E.B. Epidermolysis bullosa simplex in Scotland caused by a spectrum of keratin mutations. J. Investig. Dermatol. 2007, 127, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.; Moran, B.; Sweeney, K.; Watson, R. Cardiac arrest and dilated cardiomyopathy in a patient with epidermolysis bullosa simplex and hidradenitis suppurativa: P6480. J. Am. Acad. Dermatol. 2013, 68. [Google Scholar] [CrossRef]
- Castela, E.; Tulic, M.; Rozières, A.; Bourrat, E.; Nicolas, J.F.; Kanitakis, J.; Vabres, P.; Bessis, D.; Mazereeuw, J.; Morice-Picard, F. Epidermolysis bullosa simplex generalized severe induces a T helper 17 response and is improved by apremilast treatment. Br. J. Dermatol. 2019, 180, 357–364. [Google Scholar] [CrossRef]
- Scala, E.; Cacciapuoti, S.; Garzorz-Stark, N.; Megna, M.; Marasca, C.; Seiringer, P.; Volz, T.; Eyerich, K.; Fabbrocini, G. Hidradenitis Suppurativa: Where We Are and Where We Are Going. Cells 2021, 10, 2094. [Google Scholar] [CrossRef]
- Annicchiarico, G.; Morgese, M.G.; Esposito, S.; Lopalco, G.; Lattarulo, M.; Tampoia, M.; Bonamonte, D.; Brunetti, L.; Vitale, A.; Lapadula, G. Proinflammatory cytokines and antiskin autoantibodies in patients with inherited epidermolysis bullosa. Medicine 2015, 94. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paduano, F.; Colao, E.; Grillone, T.; Vismara, M.F.M.; Amato, R.; Nisticò, S.; Mignogna, C.; Dastoli, S.; Fabiani, F.; Zucco, R.; et al. A Familial Form of Epidermolysis Bullosa Simplex Associated with a Pathogenic Variant in KRT5. Genes 2021, 12, 1503. https://doi.org/10.3390/genes12101503
Paduano F, Colao E, Grillone T, Vismara MFM, Amato R, Nisticò S, Mignogna C, Dastoli S, Fabiani F, Zucco R, et al. A Familial Form of Epidermolysis Bullosa Simplex Associated with a Pathogenic Variant in KRT5. Genes. 2021; 12(10):1503. https://doi.org/10.3390/genes12101503
Chicago/Turabian StylePaduano, Francesco, Emma Colao, Teresa Grillone, Marco Flavio Michele Vismara, Rosario Amato, Steven Nisticò, Chiara Mignogna, Stefano Dastoli, Fernanda Fabiani, Rossella Zucco, and et al. 2021. "A Familial Form of Epidermolysis Bullosa Simplex Associated with a Pathogenic Variant in KRT5" Genes 12, no. 10: 1503. https://doi.org/10.3390/genes12101503
APA StylePaduano, F., Colao, E., Grillone, T., Vismara, M. F. M., Amato, R., Nisticò, S., Mignogna, C., Dastoli, S., Fabiani, F., Zucco, R., Trapasso, F., Perrotti, N., & Iuliano, R. (2021). A Familial Form of Epidermolysis Bullosa Simplex Associated with a Pathogenic Variant in KRT5. Genes, 12(10), 1503. https://doi.org/10.3390/genes12101503