
A Novel Variant in Superoxide Dismutase 1 Gene (p.V119M) in Als Patients with Pure Lower Motor Neuron Presentation

 ,
,  , ,
, , 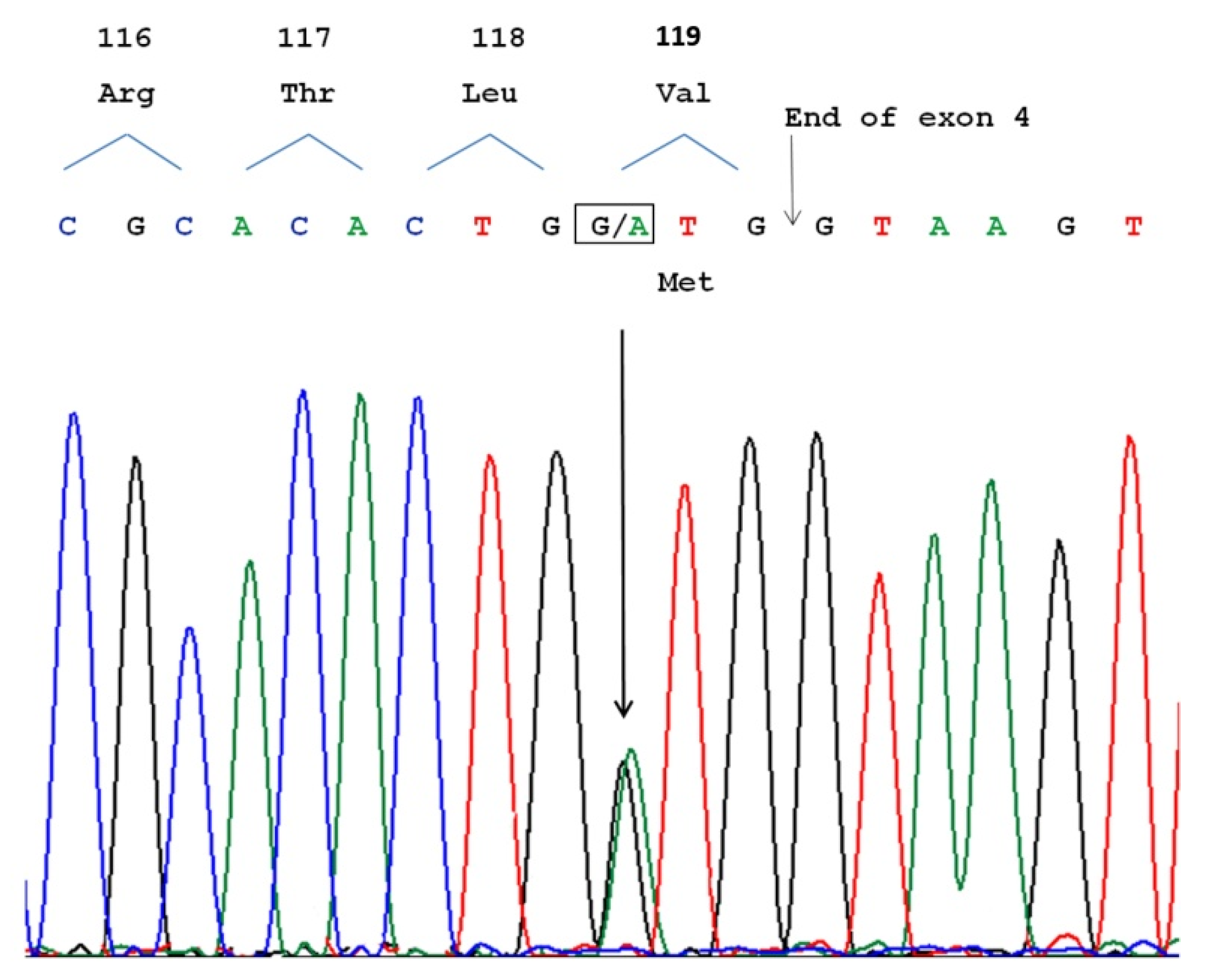{kind=link}
{kind=link}
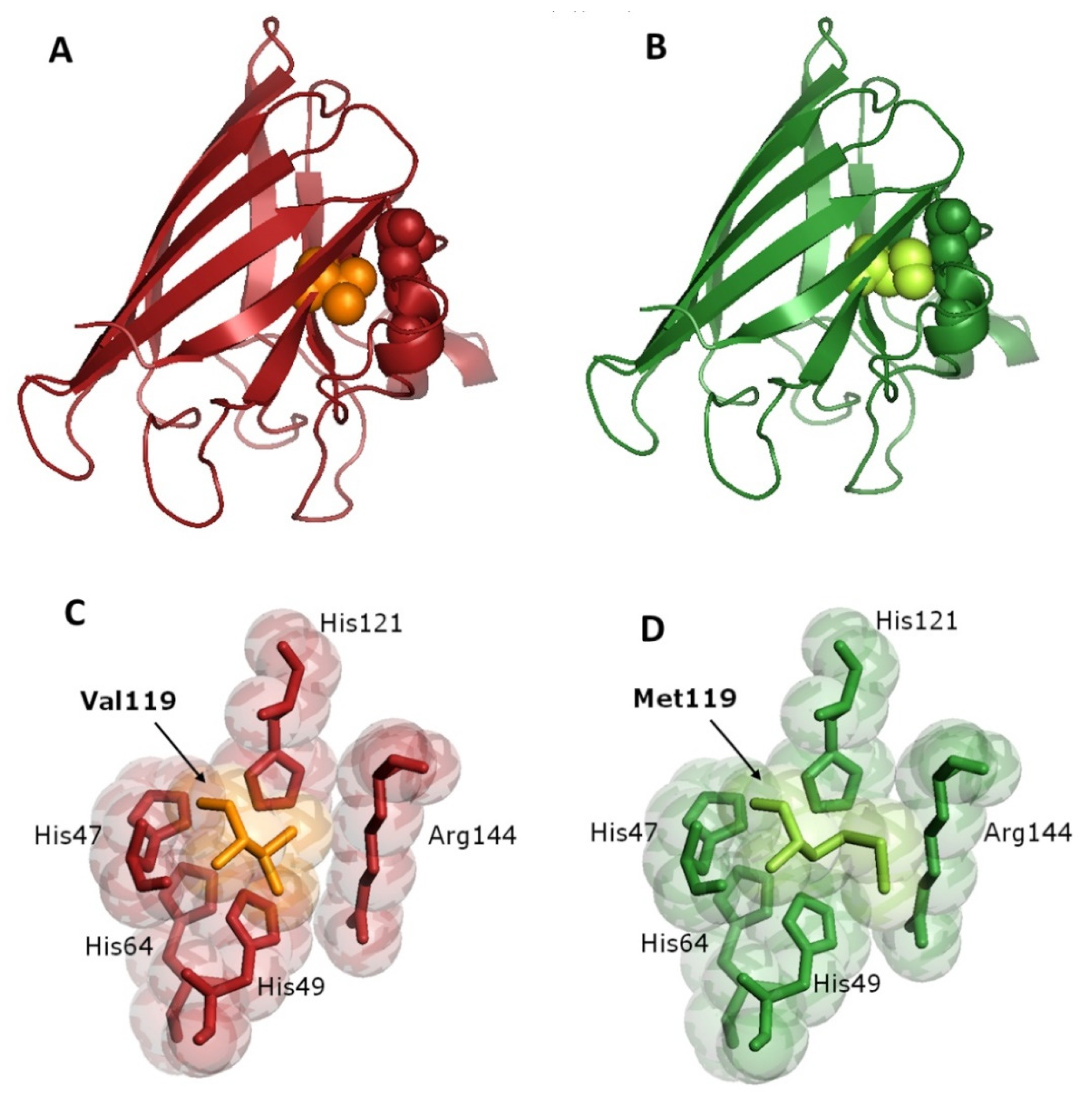{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients’ Characterization
2.2. Molecular and Bioinformatics Analysis
3. Results
3.1. Case 1
3.2. Case 2
3.3. Molecular Analysis
3.4. Bioinformatics Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gordon, P.H. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Tard, C.; Defebvre, L.; Moreau, C.; Devos, D.; Danel-Brunaud, V. Clinical features of amyotrophic lateral sclerosis and their prognostic value. Rev. Neurol. 2017, 173, 263–272. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; Vasta, R.; Brunetti, M.; Barberis, M.; Corrado, L.; D’Alfonso, S.; Bersano, E.; et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 2019, 93, e984–e994. [Google Scholar] [CrossRef]
- Bäumer, D.; Talbot, K.; Turner, M.R. Advances in motor neurone disease. J. R. Soc. Med. 2014, 107, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.; Eisen, A.; Hardiman, O.; Burrell, J.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; PARALS Study Group. Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef]
- Ravits, J.; Appel, S.; Baloh, R.H.; Barohn, R.; Brooks, B.R.; Elman, L.; Floeter, M.K.; Henderson, C.; Lomen-Hoerth, C.; Macklis, J.D.; et al. Deciphering amyotrophic lateral sclerosis: What phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 5–18. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Mathers, S.; Talman, P.; Galtrey, C.; Parkinson, M.H.; Ganesalingam, J.; Willey, E.; Ampong, M.A.; Ellis, C.M.; Shaw, C.E.; et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology 2009, 72, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Garg, N.; Park, S.B.; Vucic, S.; Yiannikas, C.; Spies, J.; Howells, J.; Huynh, W.; Matamala, J.M.; Krishnan, A.; Pollard, J.D.; et al. Differentiating lower motor neuron syndromes. J. Neurol. Neurosurg. Psychiatry 2016, 88, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Norris, F.H. Adult Progressive Muscular Atrophy and Hereditary Spinal Muscular Atrophies. In Handbook of Clinical Neurolo-gy: Diseases of the Motor System; Vinken, P.J., Bruyn, G.W., Klawans, H.L., De Jong, J.M.B.V., Eds.; Elsevier: San Diego, CA, USA, 1991; Volume 59, pp. 13–34. [Google Scholar]
- De Carvalho, M.; Scotto, M.; Swash, M. Clinical patterns in progressive muscular atrophy (PMA): A prospective study. Amyotroph. Lateral Scler. 2007, 8, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.; Heverin, M.; Doherty, M.A.; Davis, N.; Corr, E.M.; Vajda, A.; Pender, N.; McLaughlin, R.; Hardiman, O. Determining the incidence of familiality in ALS. Neurol. Genet. 2018, 4, e239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.; Pitout, I.L.; Fletcher, S.; Wilton, S.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [Green Version]
- Pardo, C.A.; Xu, Z.; Borchelt, D.R.; Price, D.L.; Sisodia, S.S.; Cleveland, D. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc. Natl. Acad. Sci. USA 1995, 92, 954–958. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.; Pu, H.; Chiu, A.; Canto, M.D.; Polchow, C.; Alexander, D.; Caliendo, J.; Hentati, A.; Kwon, Y.; Deng, H.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Bruijn, L.; Becher, M.; Lee, M.; Anderson, K.; Jenkins, N.; Copeland, N.; Sisodia, S.; Rothstein, J.; Borchelt, D.; Price, D.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.R.; Wilcox, H.M.; Flood, D.G.; Beal, M.F., Jr.; Scott, R.W.; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58. [Google Scholar] [CrossRef]
- Bali, T.; Self, W.; Liu, J.; Siddique, T.; Wang, L.H.; Bird, T.D.; Ratti, E.; Atassi, N.; Boylan, K.B.; Glass, J.D.; et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J. Neurol. Neurosurg. Psychiatry 2016, 88, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Wurster, C.D.; Ludolph, A.C. Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord. 2018, 11, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.; Cudkowicz, M.; Shaw, P.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Kimura, F.; Fujimura, C.; Ishida, S.; Nakajima, H.; Furutama, D.; Uehara, H.; Shinoda, K.; Sugino, M.; Hanafusa, T. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006, 66, 265–267. [Google Scholar] [CrossRef]
- Battistini, S.; Ricci, C.; Giannini, F.; Calzavara, S.; Greco, G.; Del Corona, A.; Mancuso, M.; Battistini, N.; Siciliano, G.; Carrera, P. G41SSOD1mutation: A common ancestor for six ALS Italian families with an aggressive phenotype. Amyotroph. Lateral Scler. 2010, 11, 210–215. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; McKenna-Yasek, D.; Sapp, P.E.; Chin, W.; Geller, B.; Hayden, D.L.; Schoenfeld, D.A.; Hosler, B.A.; Horvitz, H.R.; Brown, R.H. Epidemiology of mutations in superoxide dismutase in amyotrophic lateal sclerosis. Ann. Neurol. 1997, 41, 210–221. [Google Scholar] [CrossRef]
- Rainero, I.; Pinessi, L.; Tsuda, T.; Vignocchi, M.G.; Vaula, G.; Calvi, L.; Cerrato, P.; Rossi, B.; Bergamini, L.; McLachlan, D.R.C.; et al. SOD1 missense mutation in an Italian family with ALS. Neurology 1994, 44, 347–349. [Google Scholar] [CrossRef]
- Chio, A.; Traynor, B.J.; Lombardo, F.; Fimognari, M.; Calvo, A.; Ghiglione, P.; Mutani, R.; Restagno, G. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 2008, 70, 533–537. [Google Scholar] [CrossRef]
- Berdyński, M.; Kuźma-Kozakiewicz, M.; Ricci, C.; Kubiszewska, J.; Millecamps, S.; Salachas, F.; Łusakowska, A.; Carrera, P.; Meininger, V.; Battistini, S.; et al. Recurrent G41S mutation in Cu/Zn superoxide dismutase gene (SOD1) causing familial amyotrophic lateral sclerosis in a large Polish family. Amyotroph. Lateral Scler. 2011, 13, 132–136. [Google Scholar] [CrossRef]
- Aksoy, H.; Deng, H.; Juneja, T.; Storey, E.; Gardner, R.M.; Jacob, R.L.; Siddique, T.; Dean, G.; Elian, M.; Deng, G.; et al. A4T Mutation in the SOD1 Gene Causing Familial Amyotrophic Lateral Sclerosis. Neuroepidemiology 2003, 22, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Régal, L.; Vanopdenbosch, L.; Tilkin, P.; Bosch, L.V.D.; Thijs, V.; Sciot, R.; Robberecht, W. The G93C Mutation in Superoxide Dismutase 1. Arch. Neurol. 2006, 63, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; D’Ovidio, F.; Vasta, R.; Grassano, M.; Brunetti, M.; Barberis, M.; Corrado, L.; et al. ALS phenotype is influenced by age, sex, and genetics. Neurology 2020, 94, e802–e810. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-K.; Liu, X.; Sandner, J.; Pasmantier, M.; Andrews, J.; Rowland, L.P.; Mitsumoto, H. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology 2009, 73, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Sims, K.B.; Xin, W.W.; Kiely, R.; O’Neill, G.; Ravits, J.; Pioro, E.; Harati, Y.; Brower, R.D.; Levine, J.S.; et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: A decade of discoveries, defects and disputes. Amyotroph. Lateral Scler. 2003, 4, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kawata, A.; Kato, S.; Hayashi, M.; Takamoto, K.; Hirai, S.; Yamaguchi, S.; Komori, T.; Oda, M. Autonomic failure in ALS with a novel SOD1 gene mutation. Neurology 2000, 54, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, T.; Polak, M.; Kelly, C.; Birve, A.; Andersen, P.; Marklund, S.L.; Glass, J.D. A novel ALS SOD1 C6S mutation with implications for aggregation related toxicity and genetic counseling. Amyotroph. Lateral Scler. 2010, 12, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Kohno, S.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; Mizoguchi, K. A novel mutation (Cys6Gly) in the Cu/Zn superoxide dismutase gene associated with rapidly progressive familial amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 276, 135–137. [Google Scholar] [CrossRef]
- Morita, M.; Aoki, M.; Abe, K.; Hasegawa, T.; Sakuma, R.; Onodera, Y.; Ichikawa, N.; Nishizawa, M.; Itoyama, Y. A novel two-base mutation in the CuZn superoxide dismutase gene associated with familial amyotrophic lateral sclerosis in Japan. Neurosci. Lett. 1996, 205, 79–82. [Google Scholar] [CrossRef]
- Lindberg, M.J.; Byström, R.; Boknäs, N.; Andersen, P.M.; Oliveberg, M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc. Natl. Acad. Sci. USA 2005, 102, 9754–9759. [Google Scholar] [CrossRef] [Green Version]
- Seetharaman, S.V.; Prudencio, M.; Karch, C.; Holloway, S.P.; Borchelt, D.R.; Hart, P.J. Immature Copper-Zinc Superoxide Dismutase and Familial Amyotrophic Lateral Sclerosis. Exp. Biol. Med. 2009, 234, 1140–1154. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.-F. Superoxide dismutases: Active sites that save, but a protein that kills. Curr. Opin. Chem. Biol. 2004, 8, 162–168. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, C.; Giannini, F.; Riolo, G.; Bocci, S.; Casali, S.; Battistini, S. A Novel Variant in Superoxide Dismutase 1 Gene (p.V119M) in Als Patients with Pure Lower Motor Neuron Presentation. Genes 2021, 12, 1544. https://doi.org/10.3390/genes12101544
Ricci C, Giannini F, Riolo G, Bocci S, Casali S, Battistini S. A Novel Variant in Superoxide Dismutase 1 Gene (p.V119M) in Als Patients with Pure Lower Motor Neuron Presentation. Genes. 2021; 12(10):1544. https://doi.org/10.3390/genes12101544
Chicago/Turabian StyleRicci, Claudia, Fabio Giannini, Giulia Riolo, Silvia Bocci, Stefania Casali, and Stefania Battistini. 2021. "A Novel Variant in Superoxide Dismutase 1 Gene (p.V119M) in Als Patients with Pure Lower Motor Neuron Presentation" Genes 12, no. 10: 1544. https://doi.org/10.3390/genes12101544
APA StyleRicci, C., Giannini, F., Riolo, G., Bocci, S., Casali, S., & Battistini, S. (2021). A Novel Variant in Superoxide Dismutase 1 Gene (p.V119M) in Als Patients with Pure Lower Motor Neuron Presentation. Genes, 12(10), 1544. https://doi.org/10.3390/genes12101544