
Eukaryotic Genomes Show Strong Evolutionary Conservation of k-mer Composition and Correlation Contributions between Introns and Intergenic Regions



Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Genome Sequences
2.2. k-mer Analysis
2.3. Correlation Decomposition
3. Results
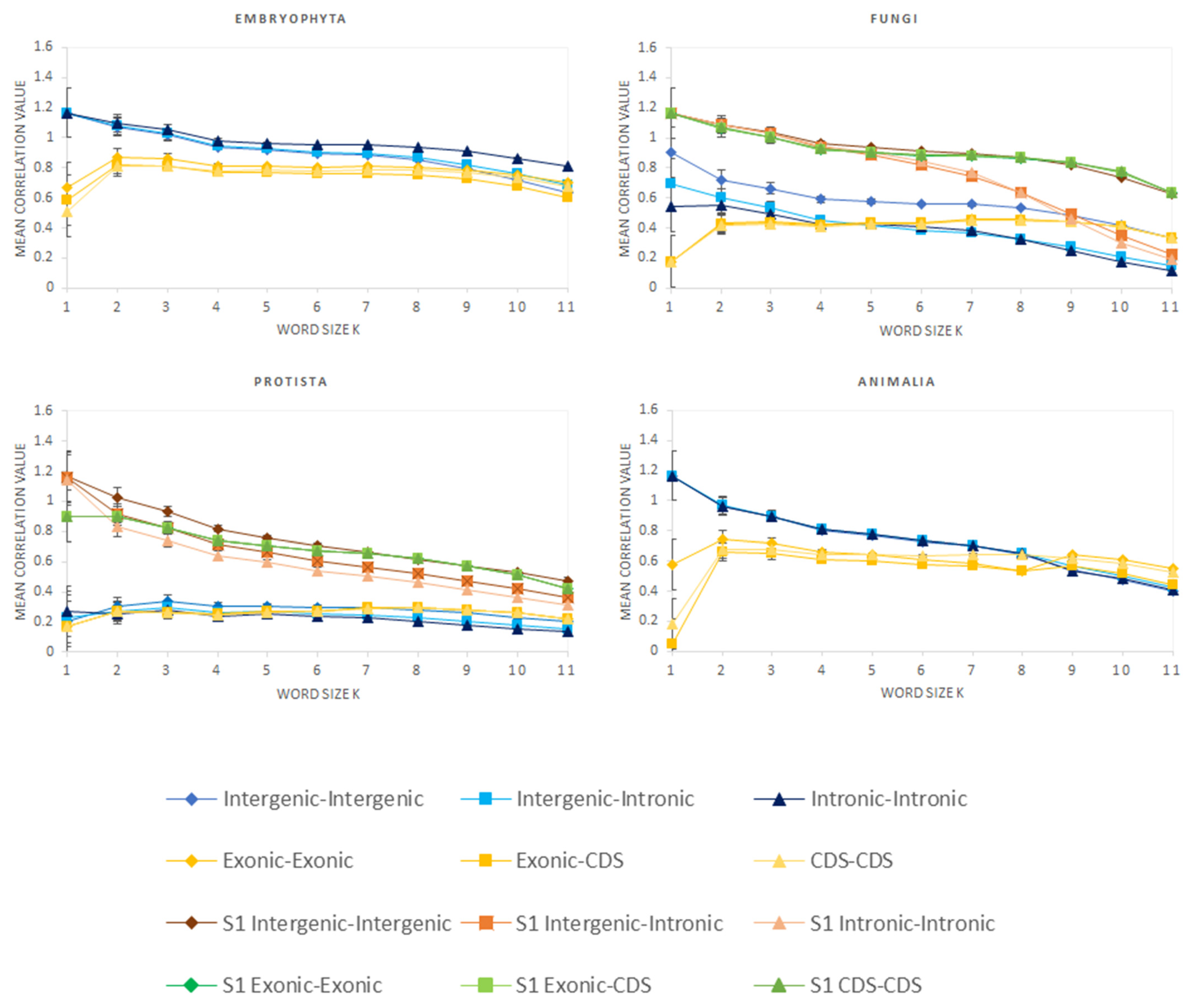
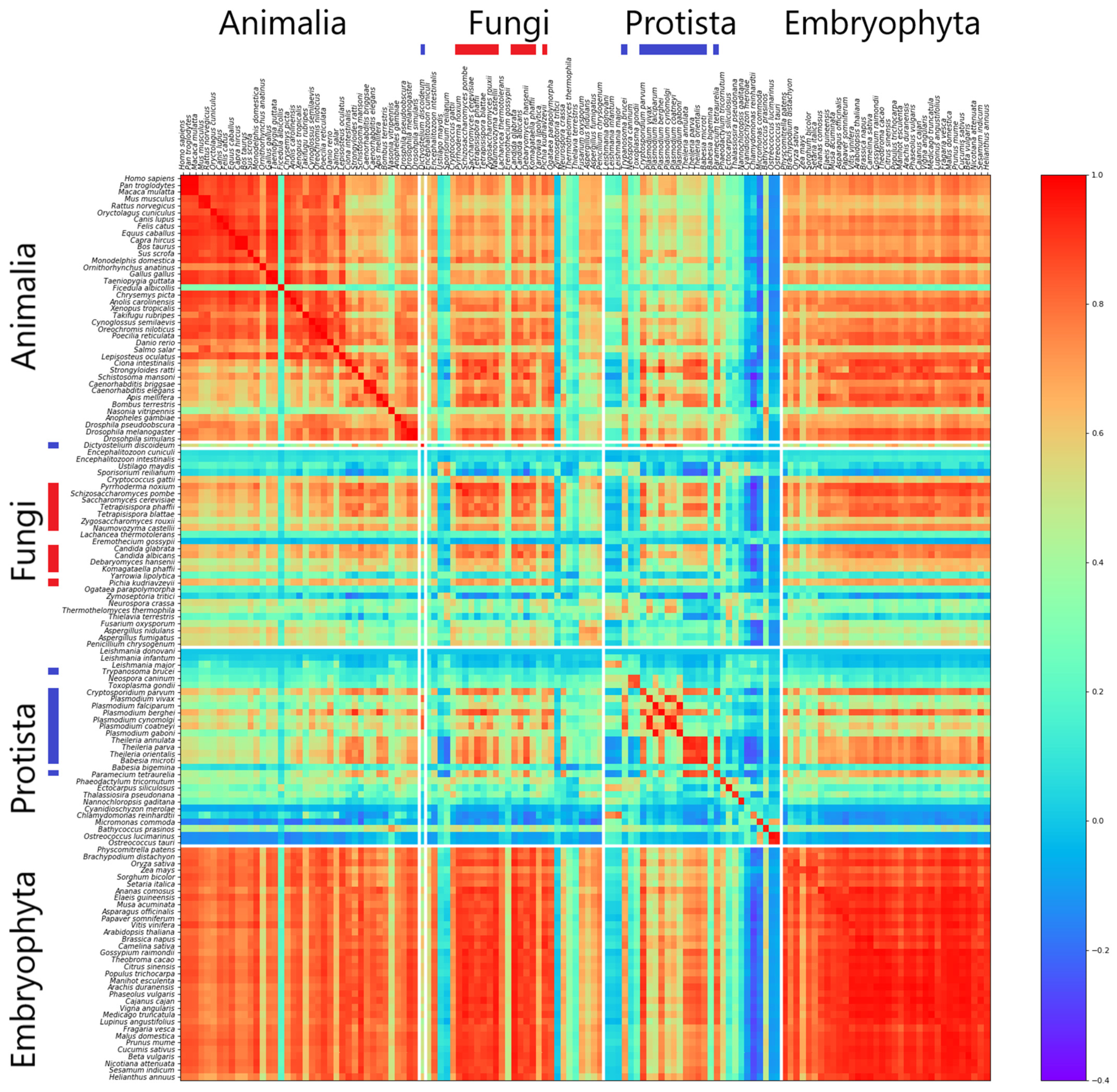
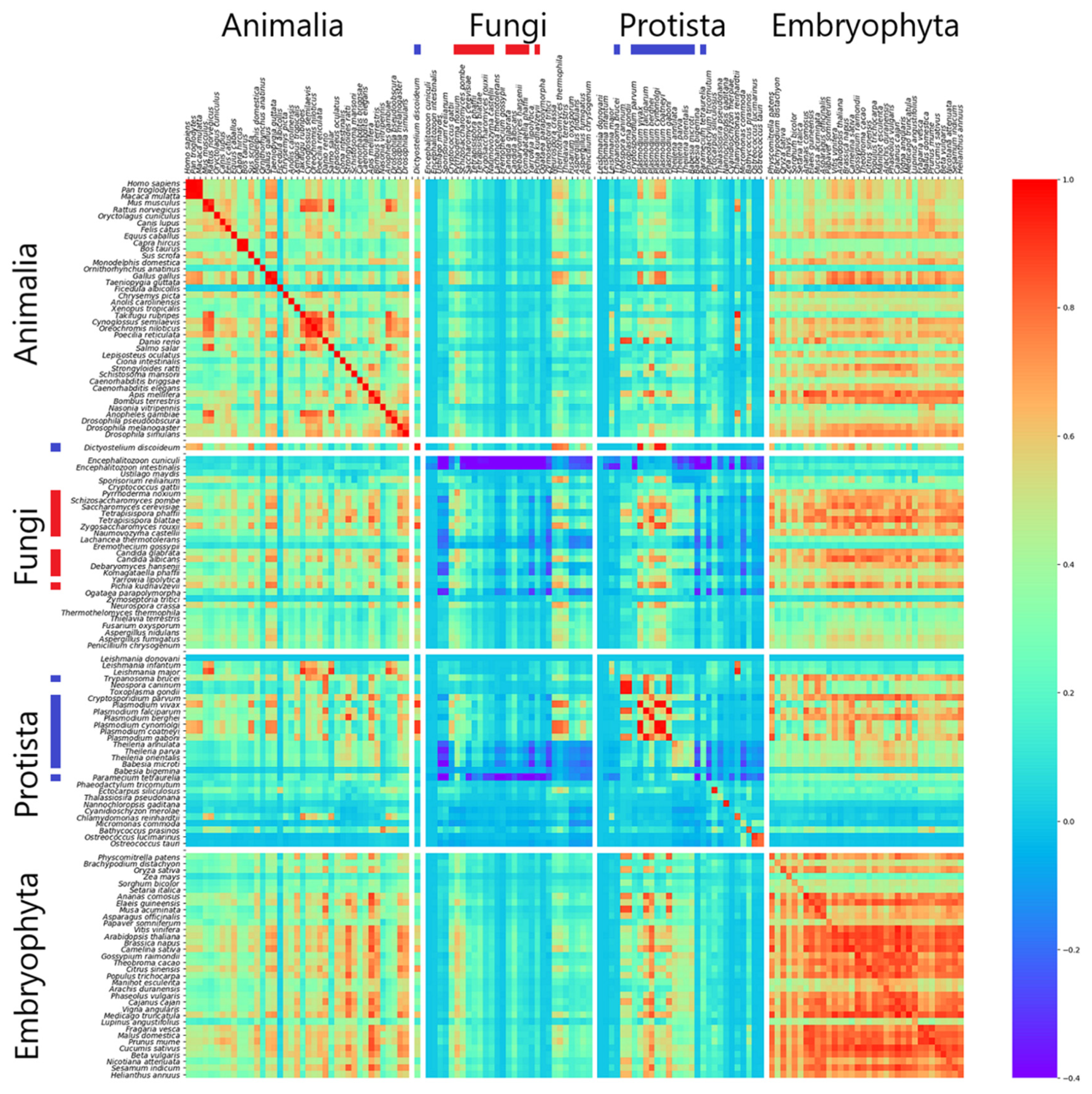
3.1. Correlation of k-mer Spectra
3.2. Abundance and Correlation Contributions of k-Words
3.3. Mismatches and Correlation Contributions of k-Words
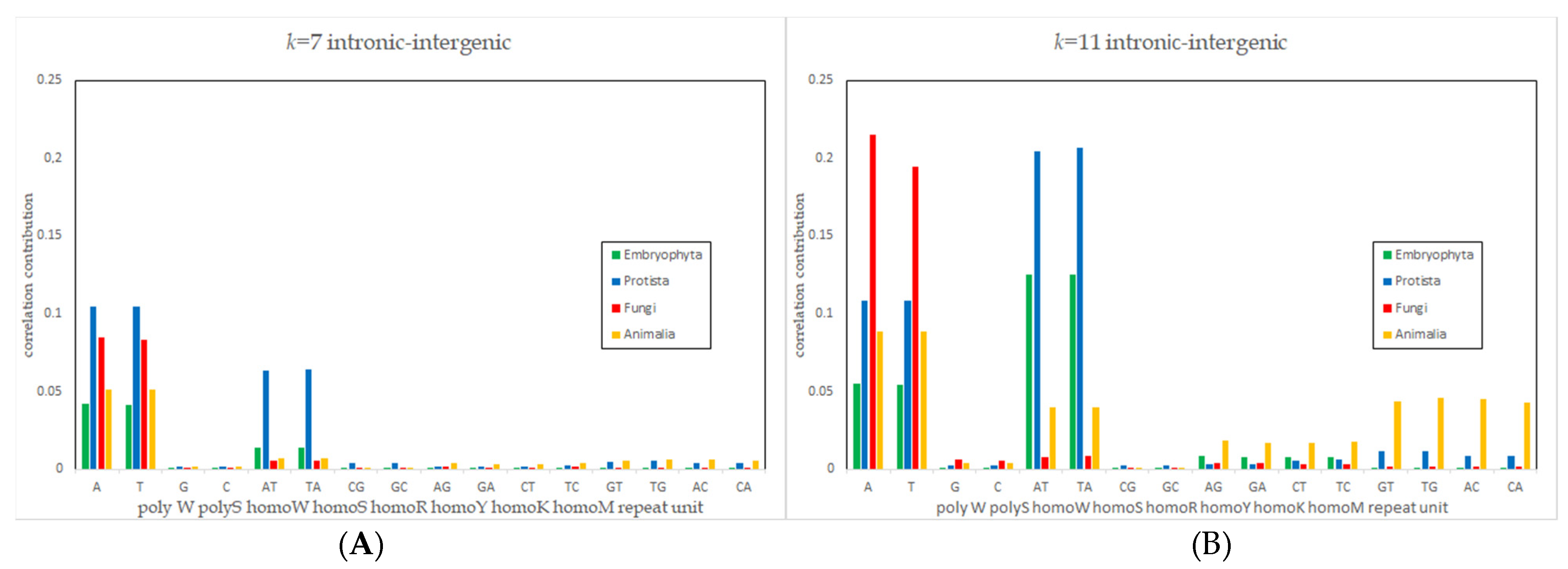
3.4. Deviation Pattern Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nirenberg, M.W.; Matthaei, J.H. The Dependence of Cell-Free Protein Synthesis in E. Coli upon Naturally Occurring or Synthetic Polyribonucleotides. Proc. Natl. Acad. Sci. USA 1961, 47, 1588–1602. [Google Scholar] [CrossRef] [Green Version]
- Ehret, C.F.; De Haller, G. Origin, Development, and Maturation of Organelles and Organelle Systems of the Cell Surface in Paramecium. J. Ultrastruct. Res. 1963, 9, 1–42. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Gregory, T.R. The Case for Junk DNA. PLoS Genet. 2014, 10, e1004351. [Google Scholar] [CrossRef] [Green Version]
- Hare, M.P. High Intron Sequence Conservation across Three Mammalian Orders Suggests Functional Constraints. Mol. Biol. Evol. 2003, 20, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Olman, V.; Xu, Y. Barcodes for Genomes and Applications. BMC Bioinform. 2008, 9, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chor, B.; Horn, D.; Goldman, N.; Levy, Y.; Massingham, T. Genomic DNA K-Mer Spectra: Models and Modalities. Genome Biol. 2009, 10, R108. [Google Scholar] [CrossRef] [Green Version]
- Francis, W.R.; Wörheide, G. Similar Ratios of Introns to Intergenic Sequence across Animal Genomes. Genome Biol. Evol. 2017, 9, 1582–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, A.; Wenz, F.; Hausmann, M.; Hildenbrand, G. Conservation of k-mer Composition and Correlation Contribution between Introns and Intergenic Regions of Animalia Genomes. Genes 2018, 9, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chan, C.X.; Ragan, M.A. Next-Generation Phylogenomics. Biol. Direct 2013, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Bultrini, E. Pentamer Vocabularies Characterizing Introns and Intron-like Intergenic Tracts from Caenorhabditis Elegans and Drosophila Melanogaster. Genes 2003, 304, 183–192. [Google Scholar] [CrossRef]
- Sievers, A.; Boesik, K.; Bisch, M.; Dreessen, C.; Riedel, J.; Froß, P.; Hausmann, M.; Hildenbrand, G. K-mer Content, Correlation, and Position Analysis of Genome DNA Sequences for the Identification of Function and Evolutionary Features. Genes 2017, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K. Note on Regression and Inheritance in the Case of Two Parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar] [CrossRef]
- Benson, D.A. GenBank. Nucleic Acids Res. 2004, 33, D34–D38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Narechania, A.; Stein, J.C.; Ware, D. A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genom. 2008, 9, 517. [Google Scholar] [CrossRef] [Green Version]
- Aggarwala, V.; Voight, B.F. An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nat. Genet. 2016, 48, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Iulio, J.; Bartha, I.; Wong, E.H.M.; Yu, H.; Lavrenko, V.; Yang, D.; Jung, I.; Hicks, M.A.; Shah, N.; Kirkness, E.F.; et al. The human noncoding genome defined by genetic diversity. Nat. Genet. 2018, 50, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Adl, S.M.; Leander, B.S.; Simpson, A.G.B.; Archibald, J.M.; Anderson, O.R.; Bass, D.; Bowser, S.S.; Brugerolle, G.; Farmer, M.A.; Karpov, S.; et al. Diversity, Nomenclature, and Taxonomy of Protists. Syst. Biol. 2007, 56, 684–689. [Google Scholar] [CrossRef] [Green Version]
- Rudner, R.; Karkas, J.D.; Chargaff, E. Separation of B. subtilis DNA into complementary strands. 3. Direct analysis. Proc. Natl. Acad. Sci. USA 1968, 60, 921–922. [Google Scholar] [CrossRef] [Green Version]
- Leander, B.S. Predatory Protists. Curr. Biol. 2020, 30, R510–R516. [Google Scholar] [CrossRef]
- Duncan, B.K.; Miller, J.H. Mutagenic Deamination of Cytosine Residues in DNA. Nature 1980, 287, 560–561. [Google Scholar] [CrossRef] [PubMed]
- Fryxell, K.J.; Zuckerkandl, E. Cytosine Deamination Plays a Primary Role in the Evolution of Mammalian Isochores. Mol. Biol. Evol. 2000, 17, 1371–1383. [Google Scholar] [CrossRef] [Green Version]
- Heinen, T.J.A.J.; Staubach, F.; Häming, D.; Tautz, D. Emergence of a New Gene from an Intergenic Region. Curr. Biol. 2009, 19, 1527–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakirev, E.S.; Ayala, F.J. Pseudogenes: Are They “Junk” or Functional DNA? Annu. Rev. Genet. 2003, 37, 123–151. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S. Evolution by Gene Duplication; Springer: New York, NY, USA, 1970; 160p. [Google Scholar]
- Spadafora, C. A LINE-1-Encoded Reverse Transcriptase-Dependent Regulatory Mechanism Is Active in Embryogenesis and Tumorigenesis. Ann. N. Y. Acad. Sci. 2015, 1341, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. Repeat Master Open 4. 02013–2015. Available online: http://www.repeatmasker.org (accessed on 27 September 2021).
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA. 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef]
- Yáñez-Cuna, J.O.; Kvon, E.Z.; Stark, A. Deciphering the transcriptional cis-regulatory code. Trends Genet. 2013, 1, 11–22. [Google Scholar] [CrossRef]
- Sela, I.; Lukatsky, D.B. DNA Sequence Correlations Shape Nonspecific Transcription Factor-DNA Binding Affinity. Biophys. J. 2011, 101, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Afek, A.; Lukatsky, D.B. Positive and Negative Design for Nonconsensus Protein-DNA Binding Affinity in the Vicinity of Functional Binding Sites. Biophys. J. 2013, 105, 1653–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, M.; Mothi, N.; Muñoz, V. Eukaryotic Transcription Factors Can Track and Control Their Target Genes Using DNA Antennas. Nat. Commun. 2020, 11, 540. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Cuna, J.O.; Arnold, C.D.; Stampfel, G.; Boryn, L.M.; Gerlach, D.; Rath, M.; Stark, A. Dissection of Thousands of Cell Type-Specific Enhancers Identifies Dinucleotide Repeat Motifs as General Enhancer Features. Genome Res. 2014, 24, 1147–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levo, M.; Segal, E. In Pursuit of Design Principles of Regulatory Sequences. Nat. Rev. Genet. 2014, 15, 453–468. [Google Scholar] [CrossRef]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-Binding Specificities of Human Transcription Factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.C.J.; Hansen, L.; Abaan, H.O.; Tullius, T.D.; Margulies, E.H. Local DNA Topography Correlates with Functional Noncoding Regions of the Human Genome. Science 2009, 324, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.J.; Dauncey, M.P.; Hunter, C.A. Sequence-Dependent DNA Structure: Dinucleotide Conformational Maps. J. Mol. Biol. 2000, 295, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Matyášek, R.; Fulneček, J.; Kovařík, A. Evaluation of DNA Bending Models in Their Capacity to Predict Electrophoretic Migration Anomalies of Satellite DNA Sequences. Electrophoresis 2013, 34, 2511–2521. [Google Scholar] [CrossRef]
- Johnson, S.; Chen, Y.-J.; Phillips, R. Poly(DA:dT)-Rich DNAs Are Highly Flexible in the Context of DNA Looping. PLoS ONE 2013, 8, e75799. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Chu, J. A Brief Review of Short Tandem Repeat Mutation. Genom. Proteom. Bioinform. 2007, 5, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Cournac, A.; Koszul, R.; Mozziconacci, J. The 3D Folding of Metazoan Genomes Correlates with the Association of Similar Repetitive Elements. Nucleic Acids Res. 2015, 44, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kingdom | Intergenic (%) | Genic (%) | Intronic (%) | Exonic (%) | CDS (%) |
|---|---|---|---|---|---|
| Embryophyta | 69.5 ± 12.06 | 29.94 ± 12.18 | 16.48 ± 6.43 | 13.01 ± 8.02 | 9.03 ± 5.13 |
| Protista #S1 | 31.15 ± 11.05 | 68.58 ± 11.40 | 6.71 ± 3.77 | 61.98 ± 11.62 | 60.83 ± 11,92 |
| Protista #S2 | 32.81 ± 14.05 | 66.95 ± 13.74 | 11.01 ± 14.65 | 55.53 ± 17.60 | 51.72 ± 18,89 |
| Fungi #S1 | 29.27 ± 9.43 | 70.72 ± 9.45 | 1.70 ± 3.89 | 68.63 ± 10.49 | 66.89 ± 10.37 |
| Fungi #S2 | 35.77 ± 14.44 | 64.21 ± 14.44 | 4.30 ± 3.24 | 59.90 ± 16.59 | 55.57 ± 19.30 |
| Animalia (56 genomes) | 46.30 ± 11.79 | 53.70 ± 11.79 | 42.10 ± 10.97 | 11.60 ± 16.67 | 7.10 ± 11.14 |
| Correlated Regions | Correlation Contribution b = 1 (E/P/F/A) in % | Correlation Contribution b = 2 (E/P/F/A) in % | Correlation Contribution b = 3 (E/P/F/A) in % | Remaining Words (E/P/F/A) in % |
|---|---|---|---|---|
| Intergenic–Intergenic | 7.81/22.78/13.31/9.62 | 3.71/12.54/3.37/4.82 | 2.03/3.68/2.80/1.54 | 86.45/61.01/80.53/89.82 |
| Intergenic–Intronic | 8.29/21.21/16.86/10.52 | 3.07/15.93/1.55/5.01 | 1.68/4.11/2.59/1.53 | 86.95/58.75/79.00/84.02 |
| Intronic–Intronic | 8.25/19.46/13.61/11.22 | 2.35/18.68/0.68/5.08 | 1.37/3.27/1.73/1.36 | 88.02/58.59/83.98/82.34 |
| Exonic–Intronic | 4.88/11.64/3.18/8.28 | 1.76/5.32/0.20/2.81 | 2.43/6.65/7.51/2.26 | 90.94/76.40/89.12/86.66 |
| Exonic–Exonic | 1.47/2.48/0.18/4.02 | 1.61/0.39/0.05/0.98 | 6.90/12.51/7.72/5.18 | 90.02/84.62/92.04/89.82 |
| Exonic–CDS | 0.08/2.26/0.10/0.41 | 0.44/0.42/0.06/0.20 | 7.88/12.49/7.59/7.17 | 91.59/84.83/92.25/92.22 |
| CDS–CDS | 0.02/2.05/0.07/0.03 | 0.14/0.32/0.05/0.09 | 8.09/12.98/7.88/8.21 | 91.74/84.66/92.00/92.67 |
| Correlated Regions | Correlation Contribution b = 1 (E/P/F/A) in % | Correlation Contribution b = 2 (E/P/F/A) in % | Correlation Contribution b = 3 (E/P/F/A) in % | Remaining Words (E/P/F/A) in % |
|---|---|---|---|---|
| Intergenic–Intergenic | 8.01/23.84/33.47/17.34 | 32.17/33.28/6.66/33.40 | 1.78/2.96/1.81/2.09 | 58.04/39.92/58.06/47.27 |
| Intergenic–Intronic | 11.04/22.07/42.09/18.51 | 28.56/47.30/3.54/32.47 | 1.45/1.61/1.61/1.84 | 58.94/29.02/52.77/47.18 |
| Intronic–Intronic | 13.45/27.16/42.81/20.45 | 22.94/35.99/2.60/32.35 | 1.07/2.56/1.25/1.54 | 62.54/34.30/53.34/45.66 |
| Exonic–Intronic | 11.20/31.21/11.71/21.31 | 16.72/9.30/0.89/21.49 | 2.70/9.71/8.17/2.37 | 69.38/49.78/79.23/54.83 |
| Exonic–Exonic | 3.73/3.36/0.29/13.37 | 12.79/0.46/0.03/8.70 | 8.66/14.90/8.26/5.70 | 74.82/81.28/91.42/72.23 |
| Exonic–CDS | 0.11/2.81/0.04/0.46 | 0.83/0.36/0.01/0.60 | 10.78/15.38/8.04/9.30 | 88.28/81.45/91.92/89.65 |
| CDS–CDS | <0.01/2.42/<0.01/0.02 | 0.04/0.17/<0.01/0.03 | 10.11/15.11/8.38/9.86 | 89.84/82.30/91.62/90.09 |
| Embryophyta | Protista | Fungi | Animalia | |||||
|---|---|---|---|---|---|---|---|---|
| k-mer Word | no mm | 1 mm | no mm | 1 mm | no mm | 1 mm | no mm | 1 mm |
| AAAAAAA | 4.16% | 11.39% | 10.47% | 15.78% | 8.48% | 14.91% | 5.10% | 11.51% |
| CCCCCCC | <0.01% | 0.03% | 0.17% | 0.36% | 0.04% | 0.08% | 0.17% | 0.20% |
| GGGGGGG | <0.01% | 0.03% | 0.16% | 0.30% | 0.04% | 0.08% | 0.15% | 0.18% |
| TTTTTTT | 4.13% | 11.34% | 10.42% | 15.72% | 8.30% | 14.78% | 5.10% | 11.55% |
| ACACACA | 0.02% | 0.12% | 0.39% | 0.56% | 0.03% | 0.13% | 0.63% | 0.88% |
| AGAGAGA | 0.10% | 0.25% | 0.18% | 0.31% | 0.12% | 0.44% | 0.38% | 0.70% |
| ATATATA | 1.35% | 2.67% | 6.35% | 9.26% | 0.53% | 1.56% | 0.68% | 1.64% |
| CACACAC | 0.01% | 0.04% | 0.36% | 0.46% | 0.02% | 0.04% | 0.50% | 0.57% |
| CGCGCGC | <0.01% | 0.07% | 0.35% | 0.48% | <0.01% | 0.06% | <0.01% | 0.08% |
| CTCTCTC | 0.06% | 0.10% | 0.18% | 0.34% | 0.09% | 0.22% | 0.30% | 0.43% |
| GAGAGAG | 0.06% | 0.10% | 0.16% | 0.29% | 0.08% | 0.21% | 0.30% | 0.43% |
| GCGCGCG | <0.01% | 0.07% | 0.35% | 0.48% | <0.01% | 0.06% | <0.01% | 0.08% |
| GTGTGTG | 0.01% | 0.04% | 0.48% | 0.61% | 0.02% | 0.04% | 0.51% | 0.58% |
| TATATAT | 1.35% | 2.67% | 6.40% | 9.33% | 0.53% | 1.56% | 0.68% | 1.63% |
| TCTCTCT | 0.10% | 0.25% | 0.20% | 0.37% | 0.13% | 0.45% | 0.38% | 0.70% |
| TGTGTGT | 0.02% | 0.12% | 0.54% | 0.75% | 0.03% | 0.12% | 0.64% | 0.89% |
| AAAAAAAAAAA | 5.48% | 7.98% | 10.86% | 11.85% | 21.48% | 25.58% | 8.84% | 10.90% |
| CCCCCCCCCCC | 0.09% | 0.10% | 0.20% | 0.28% | 0.52% | 0.57% | 0.42% | 0.45% |
| GGGGGGGGGGG | 0.09% | 0.10% | 0.20% | 0.25% | 0.59% | 0.64% | 0.41% | 0.44% |
| TTTTTTTTTTT | 5.39% | 7.88% | 10.82% | 11.83% | 19.49% | 23.69% | 8.83% | 10.89% |
| ACACACACACA | 0.09% | 0.10% | 0.81% | 0.85% | 0.14% | 0.16% | 4.48% | 4.61% |
| AGAGAGAGAGA | 0.81% | 0.84% | 0.32% | 0.37% | 0.38% | 0.43% | 1.81% | 1.91% |
| ATATATATATA | 12.54% | 12.87% | 20.48% | 21.57% | 0.75% | 0.96% | 3.95% | 4.20% |
| CACACACACAC | 0.08% | 0.09% | 0.81% | 0.85% | 0.14% | 0.15% | 4.25% | 4.36% |
| CGCGCGCGCGC | <0.01% | <0.01% | 0.22% | 0.35% | <0.01% | <0.01% | <0.01% | <0.01% |
| CTCTCTCTCTC | 0.75% | 0.78% | 0.52% | 0.57% | 0.33% | 0.37% | 1.68% | 1.79% |
| GAGAGAGAGAG | 0.78% | 0.81% | 0.31% | 0.36% | 0.37% | 0.41% | 1.70% | 1.79% |
| GCGCGCGCGCG | <0.01% | <0.01% | 0.22% | 0.36% | <0.01% | <0.01% | <0.01% | <0.01% |
| GTGTGTGTGTG | 0.08% | 0.09% | 1.14% | 1.19% | 0.12% | 0.14% | 4.32% | 4.43% |
| TATATATATAT | 12.54% | 12.87% | 20.70% | 21.73% | 0.84% | 0.86% | 3.95% | 4.19% |
| TCTCTCTCTCT | 0.78% | 0.81% | 0.59% | 0.63% | 0.34% | 0.39% | 1.78% | 1.89% |
| TGTGTGTGTGT | 0.10% | 0.11% | 1.18% | 1.24% | 0.13% | 0.15% | 4.56% | 4.68% |
| Embryophyta | Protista | Fungi | Animalia | |||
|---|---|---|---|---|---|---|
| Tandem repeat subset | #S1 | #S2 | #S1 | #S2 | ||
| Value Pattern | Value Pattern | Value Pattern | Value Pattern | Value Pattern | Value Pattern | |
| polyW | inv. compl. | inv. compl. | ** | inv. compl. | inv. compl. | inv. compl. |
| polyS | inv. compl. | inv. compl. | ** | inv. compl. | inv. compl. | inv. compl. |
| homoW | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift |
| homoS | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift |
| homoR | inv. compl. | amb. | inv. compl. | amb. | inv. compl. | inv. compl. |
| homoY | inv. compl. | amb. | * inv. compl. | * amb. | inv. compl. | inv. compl. |
| homoK | inv. compl. | amb. | shift | amb. | inv. compl. | inv. compl. |
| homoM | inv. compl. | amb. | shift | * amb. | shift | inv. compl. |
| polyW | inv. compl. | inv. compl. | inv. compl. | inv. compl. | inv. compl. | inv. compl. |
| polyS | inv. compl. | inv. compl. | inv. compl. | inv. compl. | inv. compl. | inv. compl. |
| homoW | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift |
| homoS | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift | inv. compl.=shift |
| homoR | amb. | amb. | *** inv. compl. | inv. compl. | shift | amb. |
| homoY | amb. | amb. | *** inv. compl. | inv. compl. | shift | amb. |
| homoK | amb. | amb. | shift | shift | shift | amb. |
| homoM | amb. | * amb. | shift | shift | shift | amb. |
| Embryophyta | Protista | Fungi | Animalia | |
|---|---|---|---|---|
| k-mer Word | Ratio | Ratio | Ratio | Ratio |
| AAAAAAA | 2.74 | 1.51 | 1.76 | 2.26 |
| CCCCCCC | 33.39 | 2.12 | 2.00 | 1.18 |
| GGGGGGG | 32.00 | 1.88 | 2.00 | 1.20 |
| TTTTTTT | 2.75 | 1.51 | 1.78 | 2.26 |
| ACACACA | 6.00 | 1.44 | 4.33 | 1.40 |
| AGAGAGA | 2.50 | 1.72 | 3.67 | 1.84 |
| ATATATA | 1.98 | 1.46 | 2.94 | 2.41 |
| CACACAC | 4.00 | 1.28 | 2.00 | 1.14 |
| CGCGCGC | 37.27 | 1.37 | 129.11 | 100.44 |
| CTCTCTC | 1.67 | 1.89 | 2.44 | 1.43 |
| GAGAGAG | 1.67 | 1.81 | 2.63 | 1.43 |
| GCGCGCG | 37.30 | 1.37 | 118.00 | 102.16 |
| GTGTGTG | 4.00 | 1.27 | 2.00 | 1.14 |
| TATATAT | 1.98 | 1.46 | 2.94 | 2.40 |
| TCTCTCT | 2.50 | 1.85 | 3.46 | 1.84 |
| TGTGTGT | 6.00 | 1.39 | 4.00 | 1.39 |
| AAAAAAAAAAA | 1.46 | 1.09 | 1.19 | 1.23 |
| CCCCCCCCCCC | 1.11 | 1.40 | 1.10 | 1.07 |
| GGGGGGGGGGG | 1.11 | 1.25 | 1.08 | 1.07 |
| TTTTTTTTTTT | 1.46 | 1.09 | 1.22 | 1.23 |
| ACACACACACA | 1.11 | 1.05 | 1.14 | 1.03 |
| AGAGAGAGAGA | 1.04 | 1.16 | 1.13 | 1.06 |
| ATATATATATA | 1.03 | 1.05 | 1.28 | 1.06 |
| CACACACACAC | 1.13 | 1.05 | 1.07 | 1.03 |
| CGCGCGCGCGC | 1.16 | 1.60 | 1.71 | 1.20 |
| CTCTCTCTCTC | 1.04 | 1.10 | 1.12 | 1.07 |
| GAGAGAGAGAG | 1.04 | 1.16 | 1.11 | 1.05 |
| GCGCGCGCGCG | 1.14 | 1.61 | 1.84 | 1.20 |
| GTGTGTGTGTG | 1.13 | 1.04 | 1.17 | 1.03 |
| TATATATATAT | 1.03 | 1.05 | 1.02 | 1.06 |
| TCTCTCTCTCT | 1.04 | 1.07 | 1.15 | 1.06 |
| TGTGTGTGTGT | 1.10 | 1.05 | 1.15 | 1.03 |
| Number | Sequence Type | Rule | Illustrating Pattern |
|---|---|---|---|
| 1 | Polysequences | First rank for deviations is W (three ranks possible); if S∩R=G in sequence, then into W∩R=A; if S∩Y=C in sequence, then into W∩Y=T | T in polyA, A in polyT T in polyC, A in polyG |
| Homosequences | First and second rank for deviations are W (four ranks possible); the same W base as already in the sequence with no mismatch mostly appears on the first rank | A (1.), T (2.) in (AC)n A (1.), T (2.) in (GC)n A (1.), T (2.) in (GT)n T (1.), A (2.) in (TG)n | |
| 2 | homoR/homoY and polyW | Second rank is S; if R in sequence, then S∩R=G; if Y in sequence, then S∩Y=C | T (1.), G (2.) in polyA A (1.), C (2.) in polyT A (1.), T (2.) in (AG)n T (1.), C (2.) in (CT)n |
| 3 | polyW | First and second rank changed | G (1.), T (2.) in polyA C (1.), A (2.) in polyT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sievers, A.; Sauer, L.; Hausmann, M.; Hildenbrand, G. Eukaryotic Genomes Show Strong Evolutionary Conservation of k-mer Composition and Correlation Contributions between Introns and Intergenic Regions. Genes 2021, 12, 1571. https://doi.org/10.3390/genes12101571
Sievers A, Sauer L, Hausmann M, Hildenbrand G. Eukaryotic Genomes Show Strong Evolutionary Conservation of k-mer Composition and Correlation Contributions between Introns and Intergenic Regions. Genes. 2021; 12(10):1571. https://doi.org/10.3390/genes12101571
Chicago/Turabian StyleSievers, Aaron, Liane Sauer, Michael Hausmann, and Georg Hildenbrand. 2021. "Eukaryotic Genomes Show Strong Evolutionary Conservation of k-mer Composition and Correlation Contributions between Introns and Intergenic Regions" Genes 12, no. 10: 1571. https://doi.org/10.3390/genes12101571
APA StyleSievers, A., Sauer, L., Hausmann, M., & Hildenbrand, G. (2021). Eukaryotic Genomes Show Strong Evolutionary Conservation of k-mer Composition and Correlation Contributions between Introns and Intergenic Regions. Genes, 12(10), 1571. https://doi.org/10.3390/genes12101571