
A Quantitative Proteomics Approach to Gain Insight into NRF2-KEAP1 Skeletal Muscle System and Its Cysteine Redox Regulation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Inducible Skeletal Muscle-Specific Nrf2 and Keap1 Knockouts
2.2. Mass Spectrometry-Based Proteomics
2.3. Western Blot Analyses
2.4. Differential Proteomic and Pathway Enrichment Analyses
2.5. Proteomic and Bioinformatic Analyses
3. Results
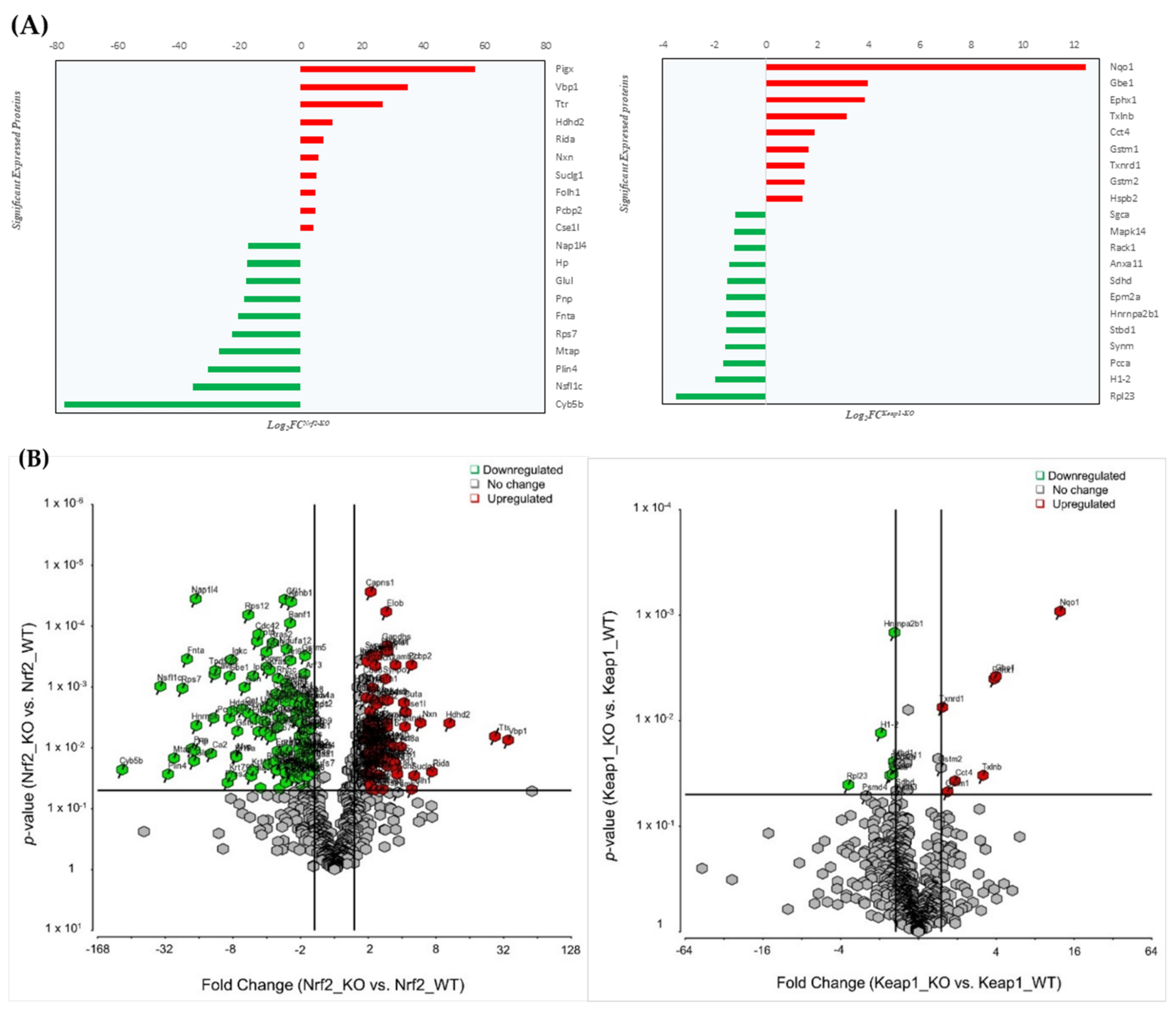
3.1. Nrf2/Keap1 Knockout Models: Mass Spectrometry Based Global Expression Analysis
3.2. MS Quantitative Analysis of Redox Regulated Proteins
3.2.1. Nrf2-KO/WT Reversibly Oxidized Redox Peptides
3.2.2. Keap1-KO/WT Reversibly Oxidized Redox Peptides
3.3. Redox Regulation Is Independent of Global Expression Change
3.4. Bioinformatics Analyses
3.4.1. Canonical Pathways
3.4.2. Networks Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Done, A.J.; Traustadottir, T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016, 10, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.; Pharaoh, G.; Premkumar, P.; Huseman, K.; Ranjit, R.; Kinter, M.; Szweda, L.; Kiss, T.; Fulop, G.; Tarantini, S.; et al. Nrf2 deficiency exacerbates age-related contractile dysfunction and loss of skeletal muscle mass. Redox Biol. 2018, 17, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Bose, C.; Alves, I.; Singh, P.; Palade, P.T.; Carvalho, E.; Borsheim, E.; Jun, S.R.; Cheema, A.; Boerma, M.; Awasthi, S.; et al. Sulforaphane prevents age-associated cardiac and muscular dysfunction through Nrf2 signaling. Aging Cell 2020, 19, e13261. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Kumar, V.; Vellichirammal, N.N.; Park, S.Y.; Rudebush, T.L.; Yu, L.; Son, W.M.; Pekas, E.J.; Wafi, A.M.; Hong, J.; et al. Functional, proteomic and bioinformatic analyses of Nrf2- and Keap1- null skeletal muscle. J. Physiol. 2020, 598, 5427–5451. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Holmgren, A.; Larsson, N.G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509. [Google Scholar] [CrossRef]
- Li, T.; He, S.; Liu, S.; Kong, Z.; Wang, J.; Zhang, Y. Effects of different exercise durations on Keap1-Nrf2-ARE pathway activation in mouse skeletal muscle. Free Radic. Res. 2015, 49, 1269–1274. [Google Scholar] [CrossRef]
- Horie, M.; Warabi, E.; Komine, S.; Oh, S.; Shoda, J. Cytoprotective Role of Nrf2 in Electrical Pulse Stimulated C2C12 Myotube. PLoS ONE 2015, 10, e0144835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wafi, A.M.; Hong, J.; Rudebush, T.L.; Yu, L.; Hackfort, B.; Wang, H.; Schultz, H.D.; Zucker, I.H.; Gao, L. Curcumin improves exercise performance of mice with coronary artery ligation-induced HFrEF: Nrf2 and antioxidant mechanisms in skeletal muscle. J. Appl. Physiol. 2019, 126, 477–486. [Google Scholar] [CrossRef]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Desai, B.N.; Myers, B.R.; Schreiber, S.L. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2002, 99, 4319–4324. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.S. mTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Popov, V.N.; Starkov, A.A. Crosstalk between the mTOR and Nrf2/ARE signaling pathways as a target in the improvement of long-term potentiation. Exp. Neurol. 2020, 328, 113285. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharples, A.P.; Hughes, D.C.; Deane, C.S.; Saini, A.; Selman, C.; Stewart, C.E. Longevity and skeletal muscle mass: The role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell 2015, 14, 511–523. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Hassannia, B.; Vandenabeele, P. An outline of necrosome triggers. Cell Mol. Life Sci. 2016, 73, 2137–2152. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.E.; Prola, A.; Mariot, V.; Pini, V.; Meng, J.; Hourde, C.; Dumonceaux, J.; Conti, F.; Relaix, F.; Authier, F.J.; et al. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 2018, 9, 3655. [Google Scholar] [CrossRef]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, P.A.; Duan, J.; Qian, W.J.; Marcinek, D.J. The Measurement of Reversible Redox Dependent Post-translational Modifications and Their Regulation of Mitochondrial and Skeletal Muscle Function. Front. Physiol. 2015, 6, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: Potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008, 283, 24801–24815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.E.; Kennedy, C.R.; Rippstein, P.; deKemp, R.; da Silva, J.; et al. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 2014, 289, 14812–14828. [Google Scholar] [CrossRef] [Green Version]
- Koser, F.; Loescher, C.; Linke, W.A. Posttranslational modifications of titin from cardiac muscle: How, where, and what for? FEBS J. 2019, 286, 2240–2260. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernandez, J.M. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Protein | Log2FCNrf2-KO | Log2FCKeap1-KO |
|---|---|---|---|
| Q9CXV1 | Sdhd | 1.87569 a | −1.48549 b |
| Q91ZA3 | Pcca | 1.69494 a | −1.65623 b |
| Q70IV5 | Synm | 1.67506 a | −1.58577 b |
| Q9JMH6 | Txnrd1 | −1.38643 b | 1.5322 a |
| P15626 | Gstm2 | −2.18014 b | 1.4955 a |
| Q9D6Y9 | Gbe1 | −8.56657 b | 3.97791 a |
| Accession | Protein | Log2FCNrf2-KO | Peptide |
|---|---|---|---|
| Q5FW97 | EG433182 | 1.71097 a | [R].SGETEDTFIADLVVGLCTGQIK.[T] |
| P17751 | Tpi1 | 1.33625 a | [K].VSHALAEGLGVIACIGEK.[L] |
| D3Z6E4 | Eno2 | 1.29545 a | [K].ACNCLLLK.[V] |
| P17751 | Tpi1 | 1.20501 a | [R].IIYGGSVTGATCK.[E] |
| Q546G4 | Alb | −1.08798 b | [R].LPCVEDYLSAILNR.[V] |
| P16125 | Ldhb | −1.17278 b | [R].VIGSGCNLDSAR.[F] |
| Q546G4 | Alb | −1.20815 b | [K].AADKDTCFSTEGPNLVTR.[C] |
| K3W4Q8 | Bsg | −1.23564 b | [R].SGEYSCIFLPEPVGR.[S] |
| Q9QUH0 | Glrx | −1.32358 b | (-).MAQEFVNCK.[I] |
| Q7TMW3 | Bgn | −1.36098 b | [R].VVQCSDLGLK.[T] |
| O08677 | Kng1 | −1.38058 b | [R].ENEFFIVTQTCK.[I] |
| P08249 | Mdh2 | −1.58447 b | [K].GCDVVVIPAGVPR.[K] |
| Q91X72 | Hpx | −1.69061 b | [K].VNSILGCSQ.(-) |
| Q3TV47 | Atp1b1 | −1.81841 b | [K].YNPNVLPVQCTGK.[R] |
| Q91X72 | Hpx | −2.73355 b | [R].DYFVSCPGR.[G] |
| Accession | Protein | Log2FCKeap1-KO | Peptide |
|---|---|---|---|
| Q9QUH0 | Glrx | 2.68731 a | (-).MAQEFVNCK.[I] |
| Q9CZU6 | Cs | −1.34429 b | [R].GYSIPECQK.[LM] |
| Q3TG92 | Actc1 | 1.99585 a | [R].CPETLFQPSFIGMESAGIHETTYNSIMK.[C] |
| D3Z1U9 | Ndufv1 | −1.42557 b | [K].NACGSDYDFDVFVVR.[G] |
| A0A0F7QZE4 | HC | −1.87462 b | [K].CPAPNLEGGPSVFIFPPNIK.[D] |
| E9Q1X8 | Cacna2d1 | 1.18133 a | [K].SYDYQSVCDPGAAPK.[Q] |
| Q3TG92 | Actc1 | 1.89048 a | [R].CPETLFQPSFIGMESAGIHETTYNSIMK.[C] |
| D3Z041 | Acsl1 | −1.59049 b | [R].GIQVSNNGPCLGSR.[K] |
| Q3TID4 | Psap | −1.39122 b | [K].SLPCDICK.[T] |
| P17751 | Tpi1 | −1.79961 b | [R].IIYGGSVTGATCK.[E] |
| Q91X72 | Hpx | −1.44668 b | [K].VNSILGCSQ.(-) |
| P17751 | Tpi1 | −1.17728 b | [R].IIYGGSVTGATCK.[E] |
| P10605 | Ctsb | −1.52989 b | [R].DQGSCGSCWAFGAVEAISDR.[T] |
| A0A1D5RLD8 | Gm10358 | −1.36198 b | [K].IVSNASCTTNCLAPLAK.[V] |
| A0A0U1RQ96 | Actg2 | 1.45841 a | [R].CPETLFQPSFI.(-) |
| P16125 | Ldhb | −1.63343 b | [R].VIGSGCNLDSAR.[F] |
| Q7TMW3 | Bgn | −1.23957 b | [R].VVQCSDLGLK.[T] |
| Q3TVI8 | Pbxip1 | −1.14557 b | [R].LQGLEANCVR.[G] |
| Q546G4 | Alb | −1.14942 b | [K].AADKDTCFSTEGPNLVTR.[C] |
| Accession | Protein | Log2FCNrf2-KO | Log2FCKeap1-KO | Peptides |
|---|---|---|---|---|
| Q9QUH0 | Glrx | −1.32358 b | 2.68731 a | (-).MAQEFVNCK.[I] |
| P17751 | Tpi1 | 1.20501 a | −1.79961 b | [R].IIYGGSVTGATCK.[E] |
| Q91X72 | Hpx | −1.69061 b | −1.44668 b | [K].VNSILGCSQ.(-) |
| P16125 | Ldhb | −1.17278 b | −1.63343 b | [R].VIGSGCNLDSAR.[F] |
| Q7TMW3 | Bgn | −1.36098 b | −1.23957 b | [R].VVQCSDLGLK.[T] |
| Q546G4 | Alb | −1.20815 b | −1.14942 b | [K].AADKDTCFSTEGPNLVTR.[C] |
| Expression | Accession | Protein | p-Value | Log2FCNrf2-KO |
|---|---|---|---|---|
| Global | P16125 | Ldhb | 0.389251 | −1.43312 b |
| Redox | P16125 | Ldhb | 0.0241312 * | −1.17278 b |
| Global | Q9QUH0 | Glrx | 0.313294 | 1.33082 a |
| Redox | Q9QUH0 | Glrx | 0.0174908 * | −1.32358 b |
| Global | P17751 | Tpi1 | 0.350314 | −1.2886 b |
| Redox | P17751 | Tpi1 | 0.0021633 * | 1.20501 a |
| Global | O08677 | Kng1 | 0.0371664 * | −2.70707 b |
| Redox | O08677 | Kng1 | 0.023165 * | −1.38058 b |
| Global | P08249 | Mdh2 | 0.249345 | 1.48018 a |
| Redox | P08249 | Mdh2 | 0.000200143 * | −1.58447 b |
| Global | P16125 | Ldhb | 0.389161 | −1.64106 b |
| Redox | P16125 | Ldhb | 0.029354 * | −1.63343 b |
| Global | Q9QUH0 | Glrx | 0.737846 | −1.12477 b |
| Redox | Q9QUH0 | Glrx | 2.50 × 10−5 * | 2.68731 a |
| Global | P17751 | Tpi1 | 0.817331 | 1.12616 a |
| Redox | P17751 | Tpi1 | 0.024665 * | −1.17728 b |
| Global | Q9CZU6 | Cs | 0.996303 | 1.00105 a |
| Redox | Q9CZU6 | Cs | 0.000104 * | −1.34429 b |
| Global | Q91X72 | Hpx | 0.1457 | −1.68804 b |
| Redox | Q91X72 | Hpx | 0.011608 * | −1.44668 b |
| Global | Q3TVI8 | Pbxip1 | 0.25651 | −1.80622 b |
| Redox | Q3TVI8 | Pbxip1 | 0.04031 * | −1.14557 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu, R.; Yu, L.; Kumar, A.; Gao, L.; Kumar, V. A Quantitative Proteomics Approach to Gain Insight into NRF2-KEAP1 Skeletal Muscle System and Its Cysteine Redox Regulation. Genes 2021, 12, 1655. https://doi.org/10.3390/genes12111655
Abu R, Yu L, Kumar A, Gao L, Kumar V. A Quantitative Proteomics Approach to Gain Insight into NRF2-KEAP1 Skeletal Muscle System and Its Cysteine Redox Regulation. Genes. 2021; 12(11):1655. https://doi.org/10.3390/genes12111655
Chicago/Turabian StyleAbu, Rafay, Li Yu, Ashok Kumar, Lie Gao, and Vikas Kumar. 2021. "A Quantitative Proteomics Approach to Gain Insight into NRF2-KEAP1 Skeletal Muscle System and Its Cysteine Redox Regulation" Genes 12, no. 11: 1655. https://doi.org/10.3390/genes12111655
APA StyleAbu, R., Yu, L., Kumar, A., Gao, L., & Kumar, V. (2021). A Quantitative Proteomics Approach to Gain Insight into NRF2-KEAP1 Skeletal Muscle System and Its Cysteine Redox Regulation. Genes, 12(11), 1655. https://doi.org/10.3390/genes12111655