1. Introduction
Malaria remains an important public health problem in several countries of tropical and subtropical regions of the world. In 2019, the disease caused an estimated 229 million clinical cases and around 409,000 deaths worldwide [
1]. Among the
Plasmodium species causing malaria in humans,
Plasmodium vivax is the most widely distributed and prevalent outside of Africa [
2]. In Brazil, endemic regions are restricted to the Legal Amazon, a region that currently accounts for the majority (>99%) of the countrywide malaria burden [
3] and where
P. vivax is predominant, with approximately 90% of the reported cases [
4]. Several exclusive features of
P. vivax biology, including the dormant liver stage, make it more resistant than other
Plasmodium species to malaria elimination [
5]. Thus,
P. vivax presents a difficult obstacle to malaria elimination in endemic countries [
6]. Therefore, it is very important to develop new methods and intervention strategies to block or reduce this transmission.
The complex life cycle of the
Plasmodium includes an erythrocytic phase that is responsible for the clinical symptoms of malaria [
7]. In this phase,
P. vivax preferentially invades reticulocytes [
8] in a process that occurs by sequential multiple molecule interactions, with each step mediated by antigens belonging to different protein families present on the merozoite surface and its apical organelles (i.e., micronemes and rhoptries) [
9], which interact with a series of specific receptors on the erythrocyte surface to complete the invasion process [
10]. The Cysteine-Rich Protective Antigen (CyRPA) is localized in the micronemes and is involved in the invasion process of merozoites into erythrocytes [
11]. In
Plasmodium falciparum, studies with the PfCyRPA protein have already demonstrated its inhibitory role in the invasion of merozoites both in vitro and in vivo, suggesting its potential as a candidate for asexual blood phase vaccine [
12,
13,
14]. However, data demonstrating the potential of
P. vivax CyRPA (PvCyRPA) as a vaccine candidate are still scarce and conflicting. França and collaborators demonstrated that antibodies against PvCyRPA are strongly related to protection. Interestingly, the protective effect of antibodies directed against PvCyRPA was higher than other proteins classically described as vaccine candidates, such as MSP-1, -3, -9 and AMA-1 [
15]. On the other hand, in vitro studies of Ndegwa et al. (2021) showed that polyclonal antibodies raised against full-length PvCyRPA did not affect
P. knowlesi growth [
16].
A vaccine able to produce antibodies that effectively prevent the invasion process after the release of merozoites into the bloodstream may decrease parasite burden, disease symptoms and, indirectly, malaria transmission [
17]. However, extensive allelic polymorphism in erythrocyte invasion pathways is known to limit the action of neutralizing antibodies against merozoite candidate vaccine antigens [
18]. Malaria parasites have abundant genetic polymorphisms, much of which have evolved to escape host immune responses and thus present a major obstacle to the development of an effective malaria vaccine [
19,
20]. The genetic diversity and population structure of
P. vivax for each candidate antigen is an important priority to the understanding of the malaria transmission dynamics [
21]. In this scenario, many studies have been proposed to investigate the global diversity of leading vaccine antigens [
22], and only one was recently addressed to the PvCyRPA [
23], which does not include Brazilian malaria-endemic areas. Therefore, to understand the potential of PvCyRPA in vaccine development, we proposed to identify
pvcyrpa gene in clinical isolates from different regions of the Brazilian Amazon and to study the potential impacts of the genetic diversity in predicted epitopes through bioinformatics tools.
4. Discussion
The invasion of the red blood cell by
Plasmodium merozoites is essential for parasite survival and proliferation. The merozoites have therefore evolved multiple pathways, using various antigenic proteins which aid in the invasion process. Among the merozoite’s invasive proteins are Cysteine-Rich Protective Antigen (CyRPA), which seems to be essential for the parasite’s life cycle during the invasion of erythrocytes and a ligand for reticulocyte invasion [
38]. The discovery of the antigen has revamped hope in the search for an effective malaria blood-stage vaccine of
P. vivax. However, one of the major obstacles to malaria vaccine development is still the low efficiency of proteins used as immunogens in inducing protection, which, in part, can be explained by genetic polymorphisms [
39]. It is important to understand the mechanisms of genetic recombination and sequence variation that represent the repertoire of polymorphic malarial surface antigens and that may help in designing vaccines [
29,
40]. The genetic diversity of these proteins in hyperendemic areas has been described as a limiting factor for the rapid acquisition of protective immunity and, consequently for the development of an effective vaccine. Furthermore, the antigenic polymorphism of
P. vivax vaccine candidates has been little discussed in unstable transmission areas such as the Brazilian endemic regions [
41]. Thus, considering that the epidemiology of malaria in Brazil presents unstable transmission and the knowledge about the genetic polymorphism of
pvcyrpa remains unknown, we aimed to identify the
pvcyrpa gene in isolates from different regions of the Brazilian Amazon and to study the potential impacts of the genetic diversity in potential B-cell epitopes.
The identification and analysis of the genetic diversity of the
pvcyrpa gene in isolates from different geographic regions of the Brazilian Amazon have not been previously studied. Considering the distance among the studied localities and the possible existence of a gene flow of Plasmodium vivax genome among the studied populations, associated with migration of people, could promote the gene flow of the parasite [
22] and impact the parasite transmission and dispersion [
42,
43]. Our first results showed that the
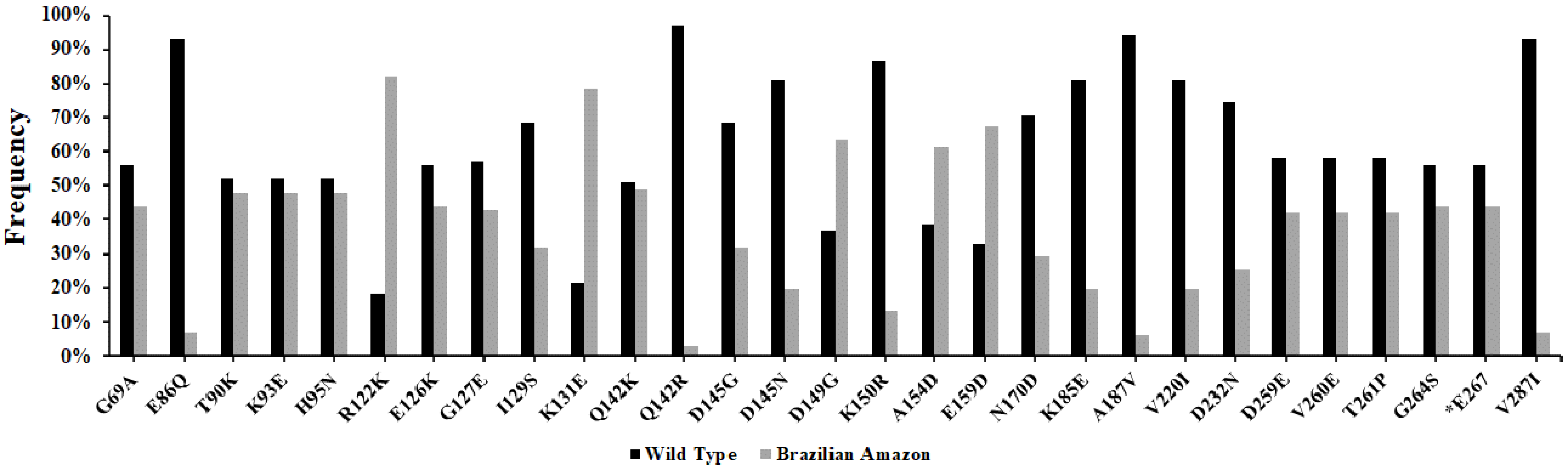
pvcyrpa gene has high genetic variability in relation to reference sequence Sal-1, presenting 27 polymorphic sites of which one was synonymous and 26 non-synonymous substitutions throughout the sequence. Among these non-synonymous substitutions, two amino acids positions—Q142 (Q142K and Q142R) and D145 (D145G and D145N)—presented one or two variants in our study areas. Overall, R122K (
N = 80%; 82%), K131E (
N = 77%; 79%), D149G (
N = 62%; 63%), A154D (
N = 60%; 61%) and E159D (
N = 66%; 67%) mutations were the most frequent in our Brazilian Amazon isolates. The analysis of the
pvcyrpa gene from the Brazilian Amazon showed that mutations have contributed to generating nucleotide and haplotype diversity. The similarity in the genetic diversity pattern suggests that similar evolutionary forces act on pvcyrpa parasites and that the structural and/or functional properties are consistent. To evade the immune response, genes encoding for antigenic proteins accumulate non-synonymous mutations, which leads to an increase in diversity. In this study, the
pvcyrpa gene presented non-synonymous mutation accumulation in parasites of different regions, mainly in exon-1. Significant positive values of Tajima’s D indicate balancing selection and population bottlenecks, while negative values suggest the presence of purifying selection or population expansion [
34]. Exon-1 had significant positive values in Cruzeiro do Sul, Guajará and Manaus for the Tajima’s Test (TjD) as well as in Manaus at exon-2. The results suggest that polymorphism at pvcyrpa exon-1 is generated by mutation and recombination, and is probably maintained by positive balancing selection pressure, which might represent an evolutionary advantage to the parasite. Exon-1 codes for highly variant domains exposed on the surface of infected red blood cells (RBCs), while exon-2 codes for the more conserved segment [
29]. Furthermore, the level of genetic diversity in blood-stage antigens seems to be associated with the degree of exposure to the immune system [
29,
44].
P. vivax biological and genetic characteristics, host immunity, and local vectors may contribute to their different patterns of demographic expansion [
45]. Some discrete
P. vivax lineages can remain stable across time in one of the areas with the highest malaria transmission in the Americas. Relapses can account for some clonal persistence because
P. vivax strains are repeatedly reintroduced in the population as hypnozoites reactivate [
46]. Maybe this context can be to explain why only Mâncio Lima and Oiapoque showed no significant positive values of Tajima’s D. However, genomic epidemiology approaches can help better to reveal the complex distribution of this parasite in the Brazilian Amazon, as well as the relationships with the worldwide genetic diversity.
Moreover, it was possible to identify 50 different haplotypes of pvcyrpa gene among the 98 P. vivax field isolates from the regions that were analyzed. The haplotype network explores the parasite relationships based on the pvcyrpa gene and comprising mutations at 34 segregating sites, and confirmed the extensive genetic diversity observed in pvcyrpa sequences. All 50 haplotypes were found to be closely related, with some of which consisting of more than one sequence from the Brazilian Amazon. Regarding the pvcyrpa sequences, we observed that haplotypes Hap_1 and Hap_11 had high frequency and shared parasites from all five localities. These findings suggest a global distribution of parasites containing similar pvcyrpa genotypes. Additionally, to compare our findings with the PvCyRPA sequences around the world, we observed that it presents a similar genetic profile among the complete genomes of P. vivax available on the GenBank Database. Among a total of 31 amino acid substitutions of PvCyRPA protein observed in P. vivax sequences worldwide, 26 amino acid substitutions are also present in our isolates.
To develop an effective malaria vaccine that can work in different regions of the world, it is important to include alleles that can induce the immune response and cover the antigenic diversity of P. vivax population. Consequently, the existence of the same haplotypes in different malaria-endemic areas and similar genetic profiles worldwide in their results will be important for the rationale of malaria vaccine designs. Moreover, as the immune system could act as selective pressure and the PvCyRPA is emerging as an alternative antigen in vaccine development, we also evaluated the impact of non-synonymous polymorphisms in relation to predicted B-cell epitope sequences.
Amino acid variation was present at peptide regions potentially participating in B-cell epitopes, which supports the idea that this molecule is under selective immune pressure. The seven B-cell potential epitopes have been identified in the PvCyRPA protein, most of which are contained in conformational epitopes, which corroborates its potential as an antibody target. A characteristic of malaria blood-stage antigens is their participation in merozoite invasion and immune evasion. Immunogenicity studies and molecular modeling are essential to determine the importance of PvCyRPA as a vaccine candidate. Targeting molecules important for the
Plasmodium life cycle might be limited by their antigenic polymorphism or low immunogenicity. Molecular studies provide information about the dynamics of vaccine antigen polymorphisms that can be used to make informed decisions about which parasite alleles to include in vaccine formulations, and to evaluate accurately the efficacy of vaccines tested in malaria-endemic areas [
21]. Thus, an effective antigen vaccine should include alleles that induce host immune responses that are sufficiently broad to cover the existing antigenic diversity. Nevertheless, because of the higher genetic diversity of
P. vivax compared to
P. falciparum, generating a broad cross-reactive immune response against highly polymorphic asexual stage antigens faces even greater challenges [
47].







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}