
Sirtuins as Metabolic Regulators of Immune Cells Phenotype and Function


Abstract
:1. Introduction
2. Sirtuins Translate Metabolic Cues to Immune Responses
2.1. The Role of Sirtuins on Innate Immune Cells
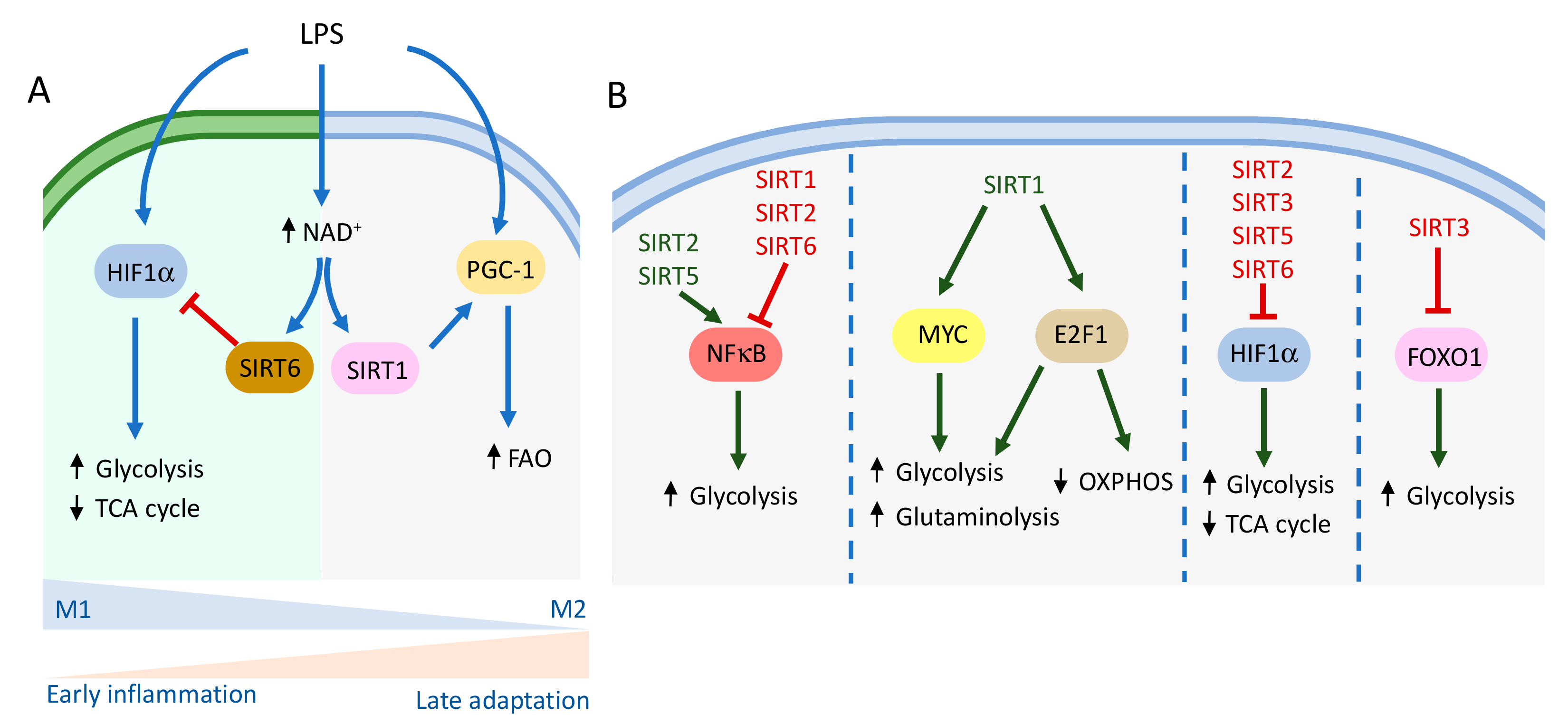
2.1.1. Macrophages
2.1.2. Dendritic Cells
2.2. The Role of Sirtuins on Adaptive Immune Cells
2.2.1. T Cells
2.2.2. B Cells
3. Sirtuins at the Crossroad of Metabolism and Immune-Related Diseases
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; He, J.; Liao, M.; Hu, M.; Li, W.; Ouyang, H.; Wang, X.; Ye, T.; Zhang, Y.; Ouyang, L. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 2019, 161, 48–77. [Google Scholar] [CrossRef]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic Shuttling of the NAD+-dependent Histone Deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Verdin, E. Interphase Nucleo-Cytoplasmic Shuttling and Localization of SIRT2 during Mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef] [Green Version]
- Osborne, B.; Cooney, G.J.; Turner, N. Are sirtuin deacylase enzymes important modulators of mitochondrial energy metabolism? Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1295–1302. [Google Scholar] [CrossRef]
- Braunstein, M.; Sobel, R.E.; Allis, C.D.; Turner, B.M.; Broach, J.R. Efficient transcriptional silencing in Saccharomyces cerevisiae requires a heterochromatin histone acetylation pattern. Mol. Cell. Biol. 1996, 16, 4349–4356. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of Insulin Secretion by SIRT4, a Mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [Green Version]
- Liszt, G.; Ford, E.; Kurtev, M.; Guarente, L. Mouse Sir2 Homolog SIRT6 Is a Nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005, 280, 21313–21320. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 Is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase. Sciences 2011, 334, 806–809. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.-Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Durso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Frescas, D.; Valenti, L.; Accili, D. Nuclear Trapping of the Forkhead Transcription Factor FoxO1 via Sirt-dependent Deacetylation Promotes Expression of Glucogenetic Genes. J. Biol. Chem. 2005, 280, 20589–20595. [Google Scholar] [CrossRef] [Green Version]
- Naiman, S.; Huynh, F.; Gil, R.; Glick, Y.; Shahar, Y.; Touitou, N.; Nahum, L.; Avivi, M.Y.; Roichman, A.; Kanfi, Y.; et al. SIRT6 Promotes Hepatic β-Oxidation via Activation of PPARα. Cell Rep. 2019, 29, 4127–4143.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Luan, H.H.; Medzhitov, R. An evolutionary perspective on immunometabolism. Sciences 2019, 363. [Google Scholar] [CrossRef]
- Pålsson-McDermott, E.M.; O’Neill, L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020, 30, 300–314. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Gu, J.; Li, T.; Chen, H.; Liu, K.; Liu, M.; Zhang, H.; Xiao, X. Inhibition of aerobic glycolysis alleviates sepsis-induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK-regulated autophagy. Int. J. Mol. Med. 2021, 47, 1. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in the development of rheumatoid arthritis. Immunol. Rev. 2020, 294, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Preyat, N.; Leo, O. Sirtuin deacylases: A molecular link between metabolism and immunity. J. Leukoc. Biol. 2013, 93, 669–680. [Google Scholar] [CrossRef]
- Vachharajani, V.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Buechler, N.L.; Woodruff, A.; Long, D.L.; Zabalawi, M.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Sirtuins and Immuno-Metabolism of Sepsis. Int. J. Mol. Sci. 2018, 19, 2738. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Vachharajani, V.; Yoza, B.K.; McCall, C.E. NAD+-dependent Sirtuin 1 and 6 Proteins Coordinate a Switch from Glucose to Fatty Acid Oxidation during the Acute Inflammatory Response. J. Biol. Chem. 2012, 287, 25758–25769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wang, X.; He, Y.; Qi, L.; Yu, L.; Xue, B.; Shi, H. The Full Capacity of AICAR to Reduce Obesity-Induced Inflammation and Insulin Resistance Requires Myeloid SIRT1. PLoS ONE 2012, 7, e49935. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nat. Cell Biol. 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 Activators Suppress Inflammatory Responses through Promotion of p65 Deacetylation and Inhibition of NF-κB Activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, T.T.; Xu, Q.; Gao, H.; Peres-Da-Silva, A.; Draper, D.W.; Fessler, M.; Purushotham, A.; Li, X. Myeloid Deletion of SIRT1 Induces Inflammatory Signaling in Response to Environmental Stress. Mol. Cell. Biol. 2010, 30, 4712–4721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imperatore, F.; Maurizio, J.; Aguilar, S.V.; Busch, C.J.; Favret, J.; Kowenz-Leutz, E.; Cathou, W.; Gentek, R.; Perrin, P.; Leutz, A.; et al. SIRT1 regulates macrophage self-renewal. EMBO J. 2017, 36, 2353–2372. [Google Scholar] [CrossRef]
- Lin, J.; Sun, B.; Jiang, C.; Hong, H.; Zheng, Y. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem. Biophys. Res. Commun. 2013, 441, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Sasso, G.L.; Menzies, K.J.; Mottis, A.; Piersigilli, A.; Perino, A.; Yamamoto, H.; Schoonjans, K.; Auwerx, J. SIRT2 Deficiency Modulates Macrophage Polarization and Susceptibility to Experimental Colitis. PLoS ONE 2014, 9, e103573. [Google Scholar] [CrossRef]
- Lee, A.S.; Jung, Y.J.; Kim, D.; Nguyen-Thanh, T.; Kang, K.P.; Lee, S.; Park, S.K.; Kim, W. SIRT2 ameliorates lipopolysaccharide-induced inflammation in macrophages. Biochem. Biophys. Res. Commun. 2014, 450, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, T.; Ciarlo, E.; Rigoni, E.; Regina, J.; Le Roy, D.; Roger, T. Dual Deletion of the Sirtuins SIRT2 and SIRT3 Impacts on Metabolism and Inflammatory Responses of Macrophages and Protects From Endotoxemia. Front. Immunol. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Chen, Y.; Jing, J.; Zhang, Y.; Liang, C.; Hao, Z.; Zhang, L. Sirtuin 3 suppresses the formation of renal calcium oxalate crystals through promoting M2 polarization of macrophages. J. Cell. Physiol. 2019, 234, 11463–11473. [Google Scholar] [CrossRef]
- Qin, K.; Han, C.; Zhang, H.; Li, T.; Li, N.; Cao, X. NAD + dependent deacetylase Sirtuin 5 rescues the innate inflammatory response of endotoxin tolerant macrophages by promoting acetylation of p65. J. Autoimmun. 2017, 81, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, K.; Xu, W.; Zhao, S.; Ye, D.; Wang, Y.; Xu, Y.; Zhou, L.; Chu, Y.; Zhang, C.; et al. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1β Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 2017, 19, 2331–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legutko, A.; Marichal, T.; Fievez, L.; Bedoret, D.; Mayer, A.; De Vries, H.; Klotz, L.; Drion, P.-V.; Heirman, C.; Cataldo, D.; et al. Sirtuin 1 Promotes Th2 Responses and Airway Allergy by Repressing Peroxisome Proliferator-Activated Receptor-γ Activity in Dendritic Cells. J. Immunol. 2011, 187, 4517–4529. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, S.-M.; Gao, B.; Zhang, J.; Fang, D. Histone Deacetylase Sirtuin 1 Deacetylates IRF1 Protein and Programs Dendritic Cells to Control Th17 Protein Differentiation during Autoimmune Inflammation. J. Biol. Chem. 2013, 288, 37256–37266. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Bi, Y.; Xue, L.; Zhang, Y.; Yang, H.; Chen, X.; Lu, Y.; Zhang, Z.; Liu, H.; Wang, X.; et al. Dendritic cell SIRT1–HIF1α axis programs the differentiation of CD4+ T cells through IL-12 and TGF-β1. Proc. Natl. Acad. Sci. USA 2015, 112, E957–E965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogoi, M.; Chandra, K.; Sarikhani, M.; Ramani, R.; Sundaresan, N.R.; Chakravortty, D. Correction: Salmonella escapes adaptive immune response via SIRT2 mediated modulation of innate immune response in dendritic cells. PLoS Pathog. 2020, 16, e1008345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, G.; Benzi, A.; Sturla, L.; Marubbi, D.; Frumento, D.; Spinelli, S.; Abbotto, E.; Ivaldi, F.; Von Holtey, M.; Murone, M.; et al. Sirt6 inhibition delays the onset of experimental autoimmune encephalomyelitis by reducing dendritic cell migration. J. Neuroinflamm. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lee, S.-M.; Shannon, S.; Gao, B.; Chen, W.; Chen, A.; Divekar, R.; McBurney, M.W.; Braley-Mullen, H.; Zaghouani, H.; et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J. Clin. Investig. 2009, 119, 3048–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bi, Y.; Chen, X.; Li, C.; Li, Y.; Zhang, Z.; Wang, J.; Lu, Y.; Yu, Q.; Su, H.; et al. Histone Deacetylase SIRT1 Negatively Regulates the Differentiation of Interleukin-9-Producing CD4 + T Cells. Immunity 2016, 44, 1337–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnölzer, M.; et al. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, C.; Kong, P.; Sun, H.; Sun, Z.; Bian, G.; Sun, Y.; Guo, L. Treatment with NAD + inhibited experimental autoimmune encephalomyelitis by activating AMPK/SIRT1 signaling pathway and modulating Th1/Th17 immune responses in mice. Int. Immunopharmacol. 2016, 39, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Berger, H.; Chalons, P.; Végan, F.; Humblin, E.; Boidot, R.; Rébé, C.; Derangère, V.; et al. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKα1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, M.Y.; Hull, P.A.; Fei, M.; Kwon, H.-S.; Tsou, C.-L.; Kasler, H.; Ng, C.-P.; Gordon, D.E.; Johnson, J.; Krogan, N.; et al. Metabolic reprogramming of human CD8+ memory T cells through loss of SIRT1. J. Exp. Med. 2017, 215, 51–62. [Google Scholar] [CrossRef]
- Kuroda, S.; Yamazaki, M.; Abe, M.; Sakimura, K.; Takayanagi, H.; Iwai, Y. Basic leucine zipper transcription factor, ATF-like (BATF) regulates epigenetically and energetically effector CD8 T-cell differentiation via Sirt1 expression. Proc. Natl. Acad. Sci. USA 2011, 108, 14885–14889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3 + T-regulatory cell function and allograft survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, Y.; Li, R.; Rezk, A.; Misirliyan, H.; Moore, C.; Farooqi, N.; Solis, M.; Goiry, L.G.; Junior, O.D.F.; Dang, V.D.; et al. A Novel MicroRNA-132-Surtuin-1 Axis Underlies Aberrant B-cell Cytokine Regulation in Patients with Relapsing-Remitting Multiple Sclerosis. PLoS ONE 2014, 9, e105421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, S.; Gordon, L.I. Functional characterization of NAD dependent de-acetylases SIRT1 and SIRT2 in B-Cell Chronic Lymphocytic Leukemia (CLL). Cancer Biol. Ther. 2016, 17, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Denu, R.A.; Krautkramer, K.; Grindle, K.M.; Yang, D.T.; Asimakopoulos, F.; Hematti, P.; Denu, J.M. Loss of SIRT3 Provides Growth Advantage for B Cell Malignancies. J. Biol. Chem. 2016, 291, 3268–3279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasha, F.; Mims, B.M.; Castro-Piedras, I.; Barnes, B.J.; Grisham, M.B.; Rahman, R.L.; Pruitt, K. The Versatility of Sirtuin-1 in Endocrinology and Immunology. Front. Cell Dev. Biol. 2020, 8, 1370. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.-T.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Zhao, Y.; Wu, X.; Xia, M.; Fang, M.; Iwasaki, Y.; Sha, J.; Chen, Q.; Xu, Y.; Shen, A. Interferon γ (IFN-γ) disrupts energy expenditure and metabolic homeostasis by suppressing SIRT1 transcription. Nucleic Acids Res. 2011, 40, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Kracht, M.; Müller-Ladner, U.; Schmitz, M.L. Mutual regulation of metabolic processes and proinflammatory NF-κB signaling. J. Allergy Clin. Immunol. 2020, 146, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef]
- Tornatore, L.; Thotakura, A.K.; Bennett, J.; Moretti, M.; Franzoso, G. The nuclear factor kappa B signaling pathway: Integrating metabolism with inflammation. Trends Cell Biol. 2012, 22, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.; Michishita, E.; Adler, A.; Damian, M.; Berber, E.; Lin, M.; Mccord, R.A.; Ongaigui, K.C.; Boxer, L.; Chang, H.Y.; et al. SIRT6 Links Histone H3 Lysine 9 Deacetylation to NF-κB-Dependent Gene Expression and Organismal Life Span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Barriopedro, I.; Bosch-Presegué, L.; Marazuela-Duque, A.; De La Torre, C.; Colomer, C.; Vazquez, B.N.; Fuhrmann, T.; Martínez-Pastor, B.; Lu, W.; Braun, T.; et al. SIRT6-dependent cysteine monoubiquitination in the PRE-SET domain of Suv39h1 regulates the NF-κB pathway. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gnanaprakasam, J.R.; Wang, R. MYC in Regulating Immunity: Metabolism and Beyond. Genes 2017, 8, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denechaud, P.-D.; Fajas, L.; Giralt, A. E2F1, a Novel Regulator of Metabolism. Front. Endocrinol. 2017, 8, 311. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Da, T.-T.; Bian, Z.-H.; He, Y.; Liu, M.-C.; Liu, Q.-Z.; Long, J.; Li, L.; Gao, C.-Y.; Yang, S.-H.; et al. Multi-omics analysis identifies FoxO1 as a regulator of macrophage function through metabolic reprogramming. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1 plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Kim, T.H. Fundamental role of dendritic cells in inducing Th2 responses. Korean J. Intern. Med. 2018, 33, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L. Glycolytic reprogramming by TLRs in dendritic cells. Nat. Immunol. 2014, 15, 314–315. [Google Scholar] [CrossRef]
- Lasigliè, D.; Boero, S.; Bauer, I.; Morando, S.; Damonte, P.; Cea, M.; Monacelli, F.; Odetti, P.; Ballestrero, A.; Uccelli, A.; et al. Sirt6 regulates dendritic cell differentiation, maturation, and function. Aging 2016, 8, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Dikshit, M. Metabolic Insight of Neutrophils in Health and Disease. Front. Immunol. 2019, 10, 2099. [Google Scholar] [CrossRef]
- Warren, J.L.; Maciver, N.J. Regulation of Adaptive Immune Cells by Sirtuins. Front. Endocrinol. 2019, 10, 466. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Kallies, A. Synchronizing transcriptional control of T cell metabolism and function. Nat. Rev. Immunol. 2015, 15, 574–584. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jetten, A.M.; Takeda, Y.; Slominski, A.; Kang, H.S. Retinoic acid-related Orphan Receptor γ (RORγ): Connecting sterol metabolism to regulation of the immune system and autoimmune disease. Physiol. Behav. 2019, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Camporeale, A.; Poli, V. STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R.; et al. Control of TH17/Treg Balance by Hypoxia-Inducible Factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.J.; Beekman, J.M.; Van Beekum, O.; Brenkman, A.B.; Hijnen, D.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef]
- Beier, U.H.; Wang, L.; Bhatti, T.R.; Liu, Y.; Han, R.; Ge, G.; Hancock, W.W. Sirtuin-1 Targeting Promotes Foxp3 + T-Regulatory Cell Function and Prolongs Allograft Survival. Mol. Cell. Biol. 2011, 31, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daenthanasanmak, A.; Iamsawat, S.; Chakraborty, P.; Nguyen, H.D.; Bastian, D.; Liu, C.; Mehrotra, S.; Yu, X.-Z. Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood 2019, 133, 266–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic but Exaggerated in Chronically BAFF-Exposed B Cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghirotto, B.; Terra, F.F.; Câmara, N.O.S.; Basso, P.J. Sirtuins in B lymphocytes metabolism and function. World J. Exp. Med. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Doughty, C.A.; Bleiman, B.F.; Wagner, D.J.; Dufort, F.J.; Mataraza, J.M.; Roberts, M.F.; Chiles, T.C. Antigen receptor–mediated changes in glucose metabolism in B lymphocytes: Role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 2006, 107, 4458–4465. [Google Scholar] [CrossRef] [Green Version]
- Dufort, F.J.; Gumina, M.R.; Ta, N.L.; Tao, Y.; Heyse, S.A.; Scott, D.A.; Richardson, A.D.; Seyfried, T.N.; Chiles, T.C. Glucose-dependent de Novo Lipogenesis in B Lymphocytes: A requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. J. Biol. Chem. 2014, 289, 7011–7024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequeira, J.; Boily, G.; Bazinet, S.; Saliba, S.; He, X.; Jardine, K.; Kennedy, C.; Staines, W.; Rousseaux, C.; Mueller, R.; et al. Sirt1-null mice develop an autoimmune-like condition. Exp. Cell Res. 2008, 314, 3069–3074. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Grötsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.-X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Foolad, F.; Khodagholi, F.; Javan, M. Sirtuins in Multiple Sclerosis: The crossroad of neurodegeneration, autoimmunity and metabolism. Mult. Scler. Relat. Disord. 2019, 34, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Kornberg, M.D. The immunologic Warburg effect: Evidence and therapeutic opportunities in autoimmunity. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Sciences 2018, 360, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Zhou, X.; Liu, Y.; Tan, S.; Li, Y. The Role of Sirtuin-1 in Immune Response and Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Quero, L.; Tiaden, A.N.; Hanser, E.; Roux, J.; Laski, A.; Hall, J.; Kyburz, D. miR-221-3p Drives the Shift of M2-Macrophages to a Pro-Inflammatory Function by Suppressing JAK3/STAT3 Activation. Front. Immunol. 2020, 10, 3087. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lu, Y.; Zhang, Z.; Wang, J.; Yang, H.; Liu, G. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology 2015, 145, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297, Erratum in 2019, 41, 299. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Kleszcz, R.; Paluszczak, J.; Baer-Dubowska, W. Targeting aberrant cancer metabolism—The role of sirtuins. Pharmacol. Rep. 2015, 67, 1068–1080. [Google Scholar] [CrossRef]
- Sebastian, C.; Mostoslavsky, R. The role of mammalian sirtuins in cancer metabolism. Semin. Cell Dev. Biol. 2015, 43, 33–42. [Google Scholar] [CrossRef] [Green Version]
- De-Brito, N.M.; Duncan-Moretti, J.; Da-Costa, H.C.; Saldanha-Gama, R.; Paula-Neto, H.A.; Dorighello, G.; Simões, R.L.; Barja-Fidalgo, C. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim. Biophys. Acta Bioenerg. 2020, 1867, 118604. [Google Scholar] [CrossRef]
- Liu, G.; Bi, Y.; Shen, B.; Yang, H.; Zhang, Y.; Wang, X.; Liu, H.; Lu, Y.; Liao, J.; Chen, X.; et al. SIRT1 Limits the Function and Fate of Myeloid-Derived Suppressor Cells in Tumors by Orchestrating HIF-1α–Dependent Glycolysis. Cancer Res. 2014, 74, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Immune Cell | Sirtuin | Known Function | Mechanism of Action | References |
|---|---|---|---|---|
| Macrophages | SIRT1 | Increases FAO | Activation of PGC1α | [25] |
| Regulates M2 macrophage polarization | Unknown, probably by increasing oxidative metabolism | [26] | ||
| Enforces oxidative metabolism | Inhibition of NF-κB signaling Activation of AMPK, PPARα and PGC1α | [27,28,29,30] | ||
| Repression of pro-inflammatory cytokine secretion | Inhibition of NF-κB transcriptional activity | [31] | ||
| Regulates insulin levels and glucose metabolism | Inhibition of NF-κB transcriptional activity | [31] | ||
| Regulates macrophage self-renewal | Activation of Myc and E2F1 and repression of FoxO | [32] | ||
| SIRT2 | Inhibits pro-inflammatory gene expression | Deacetylation of NF-κB p65 | [33,34] | |
| Induces pro-inflammatory gene expression | Activation of NF-κB | [35] | ||
| Inhibition of FAO and induction of glycolysis (together with SIRT3) | Induce the expression of HIF1α | [36] | ||
| SIRT3 | Regulation of M2 macrophage polarization | Blocking the translocation of FoxO1 to the nucleus | [37] | |
| SIRT5 | Promotes the inflammatory response | Promoting the acetylation of NF-κB | [38] | |
| Suppresses inflammation Inhibition of glycolysis? | Desuccinylation of PKM2 | [39] | ||
| SIRT6 | Inhibits glucose metabolism | Corepressing HIF1α Inhibiting NF-κB? | [25] | |
| Dendritic cells | SIRT1 | Promotes Th2 responses Regulation of glucose metabolism, fatty acid storage and lipid metabolism? | Inhibition of PPARγ | [40,41] |
| Promotes Th17 differentiation | Deacetylation of IRF1 and inhibition of IL-27 and IFN-β expression | [42] | ||
| Modulates IL-12 and TFG-1 secretion and the balance of Th1/Treg cells | Inhibition of HIF1α expression | [43] | ||
| SIRT2 | Regulates immune response to Salmonella | Induction of NF-κB translocation to the nucleus and NOS2 expression | [44] | |
| SIRT6 | Induces DC migration | Induction of TNF secretion | [45] | |
| T cells | SIRT1 | Reduces the production of Th1 and Th2 cytokines | Inhibition of AP-1 | [46] |
| Represses glycolytic metabolism and Th9 differentiation | Inhibition of mTOR-HIF1α axis | [47] | ||
| Promotes Th17 differentiation | RORγt deacetylation | [48] | ||
| Suppresses Th1 and Th17 differentiation | STAT3 deacetylation | [49,50] | ||
| Inhibits Treg suppressive function | Inhibition of FoxP3 expression Deacetylation of p53 | [51,52] | ||
| Inhibits CD8+ memory T cell differentiation | Activation of FoxO1 and inhibition of glycolytic metabolism | [53] | ||
| Blocks CD8+ effector T cell differentiation | Epigenetic repression of T-bet and inhibition of NAD+ and ATP production | [54] | ||
| SIRT3 | Promotes Treg suppressive function | Induction of oxidative metabolism | [55] | |
| B cells | SIRT1 | Suppresses lymphotoxin and TNF-α production | Inhibition of NF-κB? | [56] |
| Induces proliferation of CLL cells | Unknown | [57] | ||
| SIRT2 | Induces proliferation of CLL cells | Unknown | [57] | |
| SIRT3 | Suppresses CLL | Deacetylation of IDH2 and SOD2 and inhibition of glycolysis | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortuny, L.; Sebastián, C. Sirtuins as Metabolic Regulators of Immune Cells Phenotype and Function. Genes 2021, 12, 1698. https://doi.org/10.3390/genes12111698
Fortuny L, Sebastián C. Sirtuins as Metabolic Regulators of Immune Cells Phenotype and Function. Genes. 2021; 12(11):1698. https://doi.org/10.3390/genes12111698
Chicago/Turabian StyleFortuny, Lídia, and Carlos Sebastián. 2021. "Sirtuins as Metabolic Regulators of Immune Cells Phenotype and Function" Genes 12, no. 11: 1698. https://doi.org/10.3390/genes12111698
APA StyleFortuny, L., & Sebastián, C. (2021). Sirtuins as Metabolic Regulators of Immune Cells Phenotype and Function. Genes, 12(11), 1698. https://doi.org/10.3390/genes12111698