
Phylogenetic Signal Dissection of Heterogeneous 28S and 16S rRNA Genes in Spinicaudata (Branchiopoda, Diplostraca)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Taxon Sampling
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Sequence Alignment and Secondary Structure Prediction
2.4. Phylogenetic Analyses
2.5. Non-Phylogenetic Signal Dissection
2.5.1. Network Analysis
2.5.2. Evaluation of Compositional Heterogeneity and Substitutional Saturation
2.6. Testing Support for Competing Hypotheses
3. Results
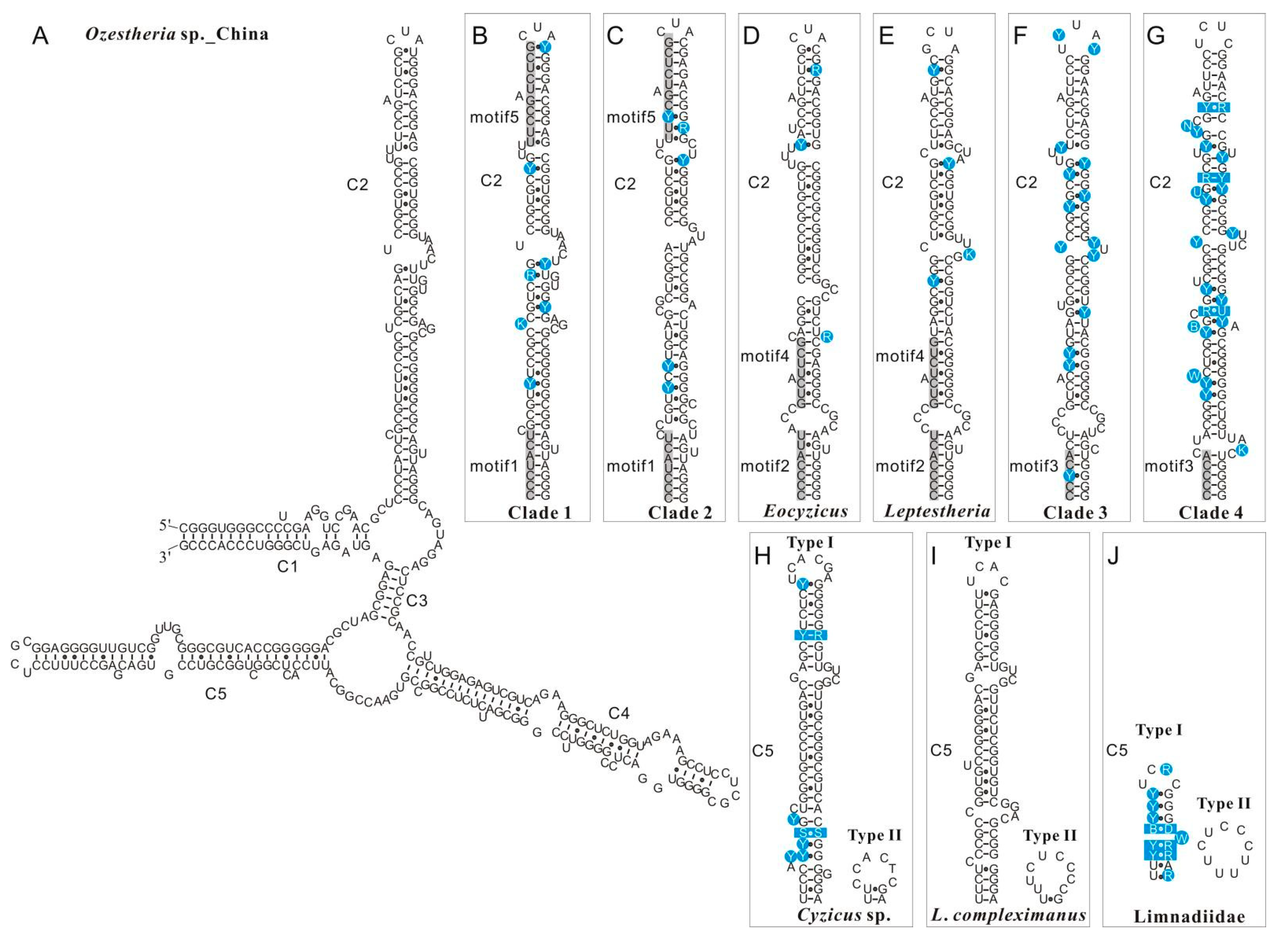
3.1. Variation and Conserved Motifs in the 28S rRNA and 16S rRNA Secondary Structure among Spinicaudata Lineages
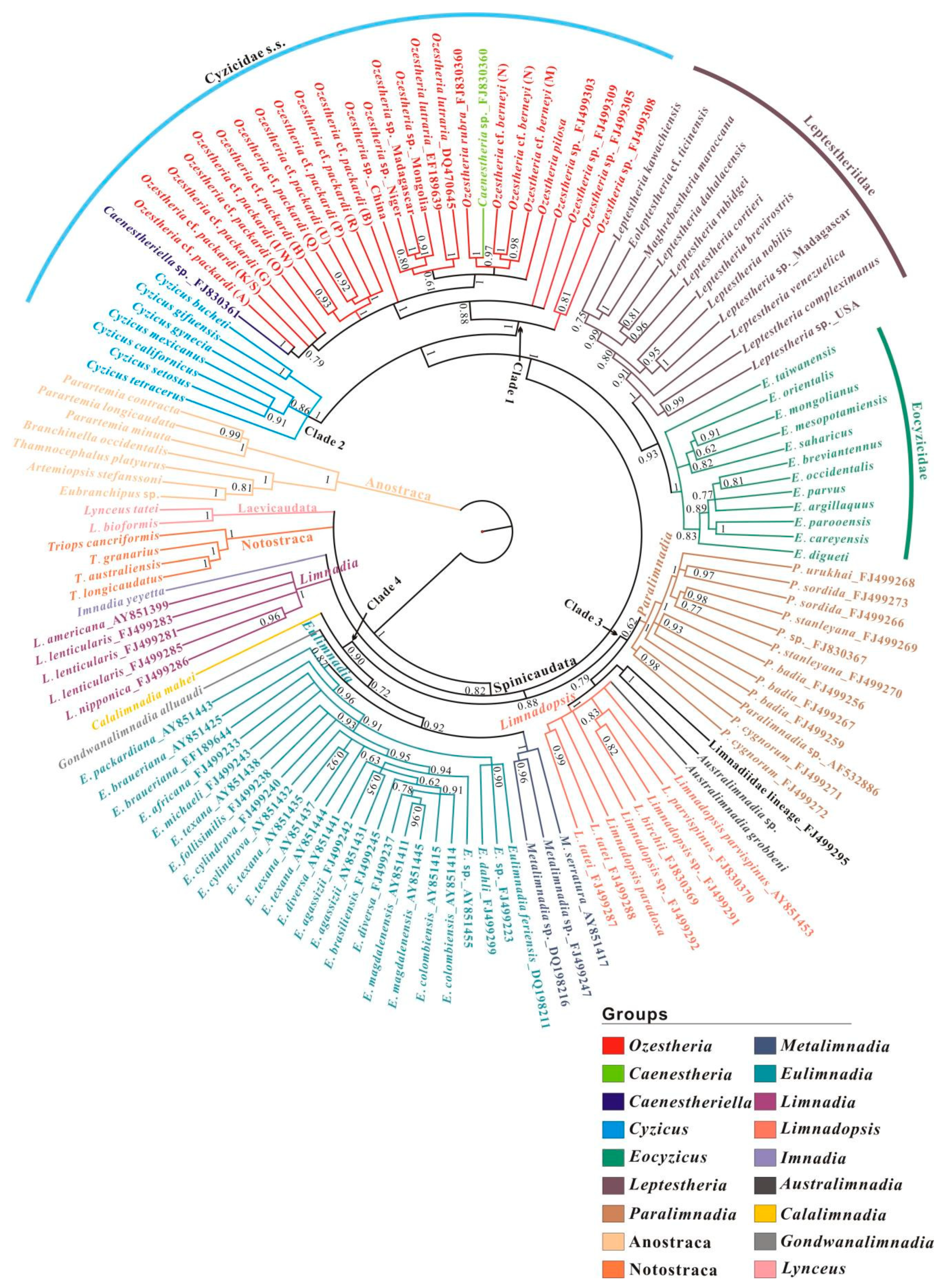
3.2. Data Exploration: Compositional Heterogeneity, Relative Homoplasy and Supernetwork
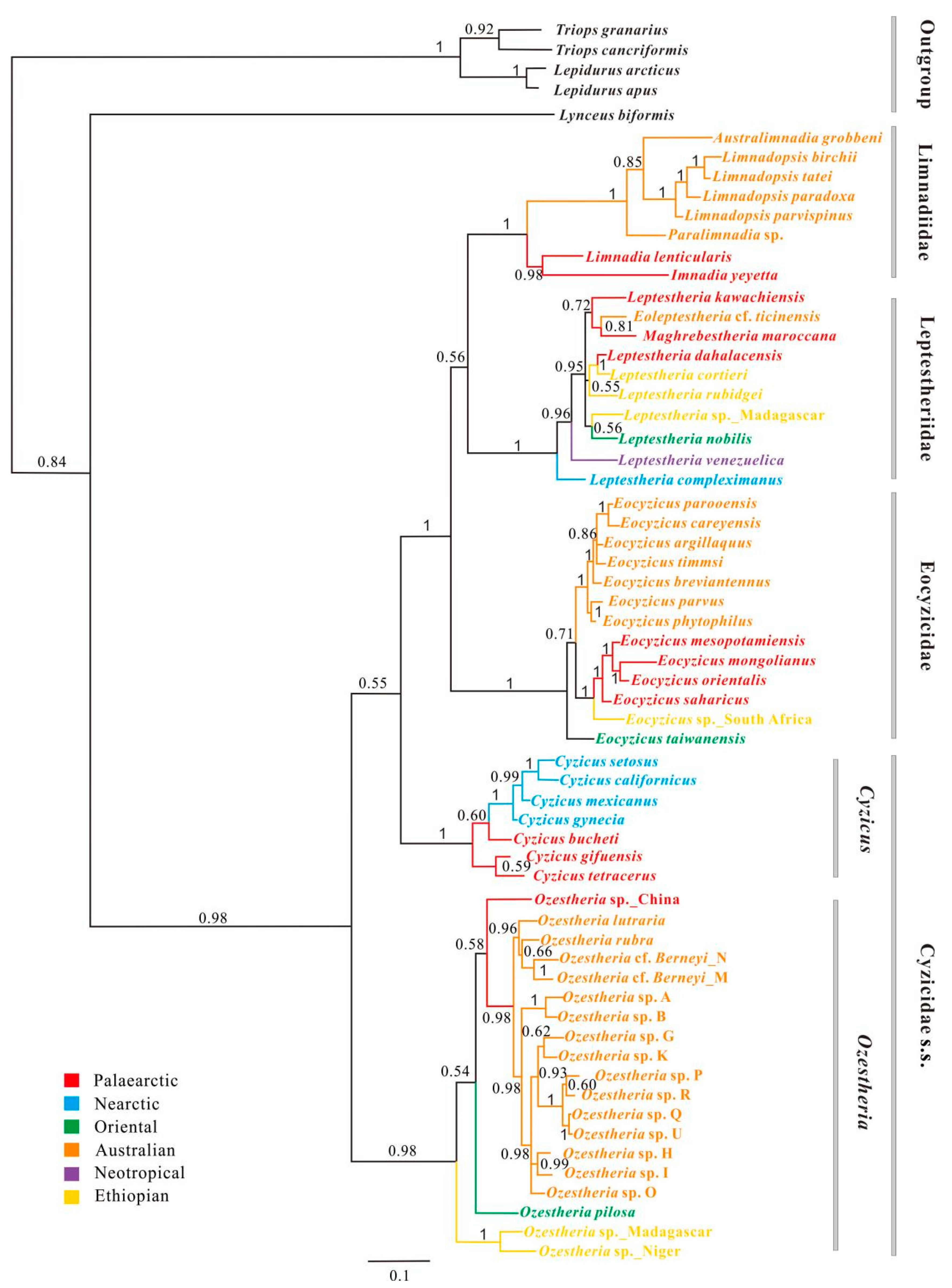
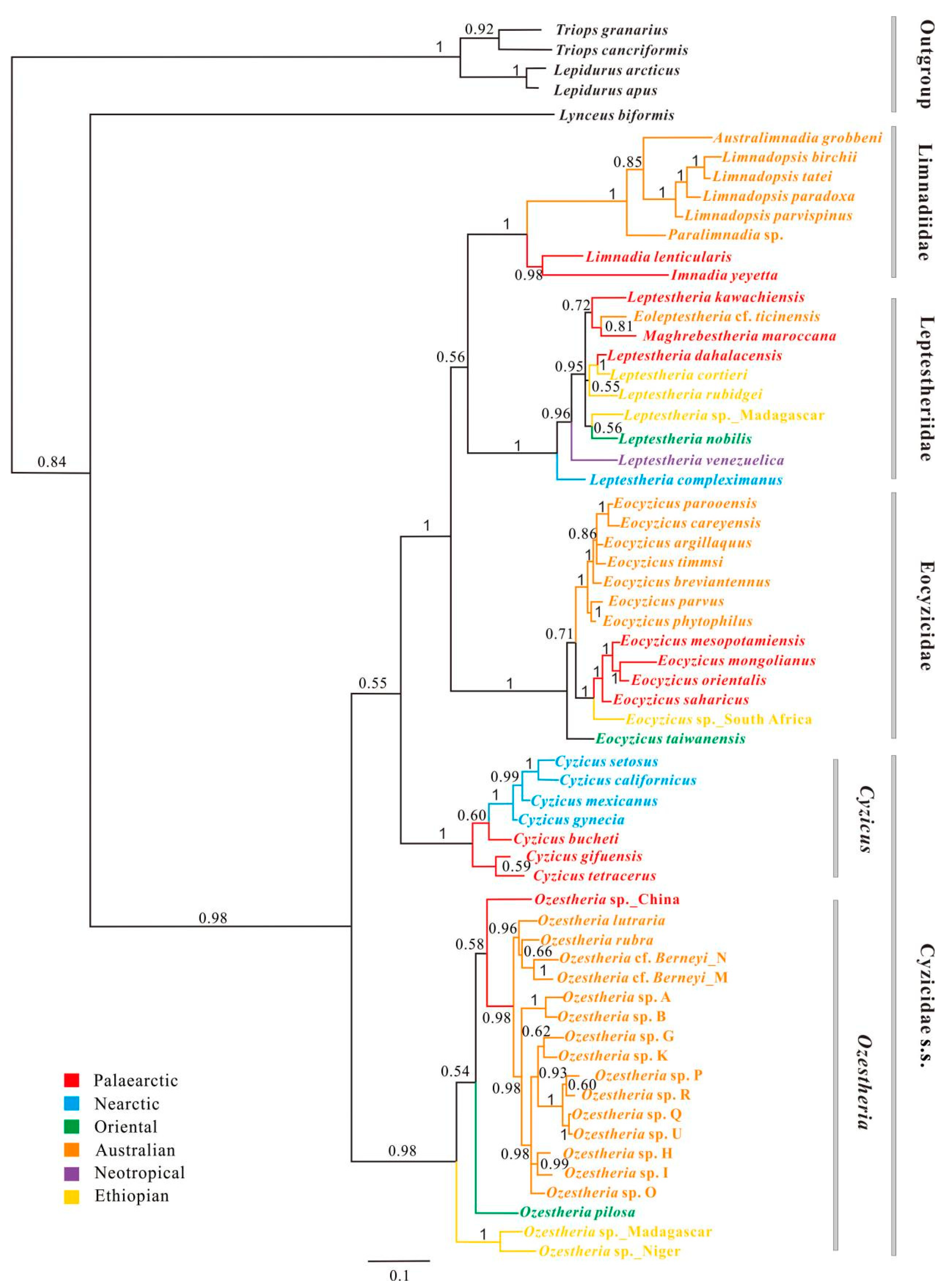
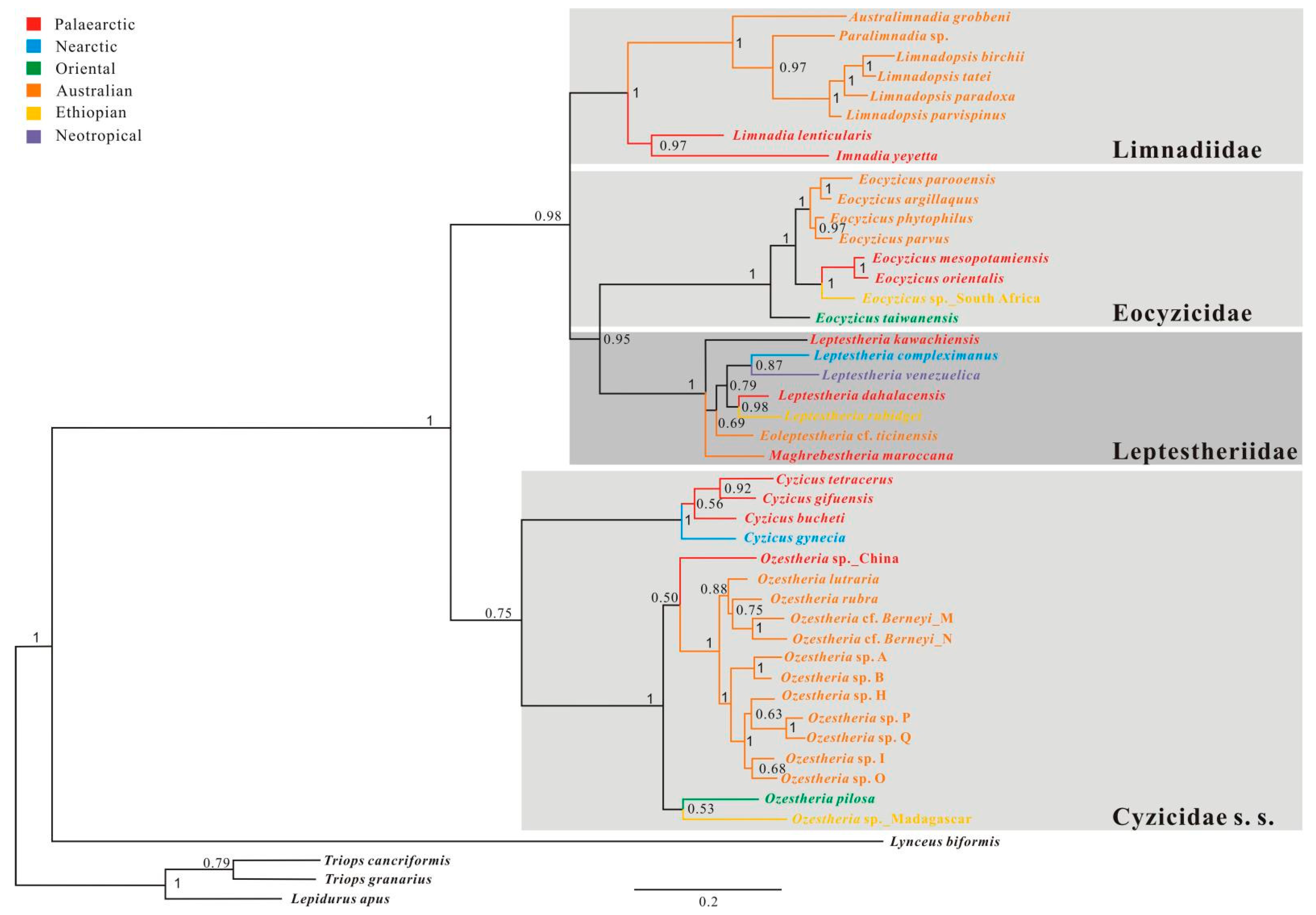
3.3. Phylogenetic Relationships among Main Clades of Spinicaudata
3.3.1. A 28S rRNA-Based Phylogeny of Spinicaudata
3.3.2. A 16S rRNA-Based Phylogeny of Spinicaudata
3.3.3. Combined 28S + 16S rRNA Analysis
3.3.4. 28S rRNA + EF1α Combined Analysis
3.3.5. 28S + 16S + EF1α + COI Combined Analysis
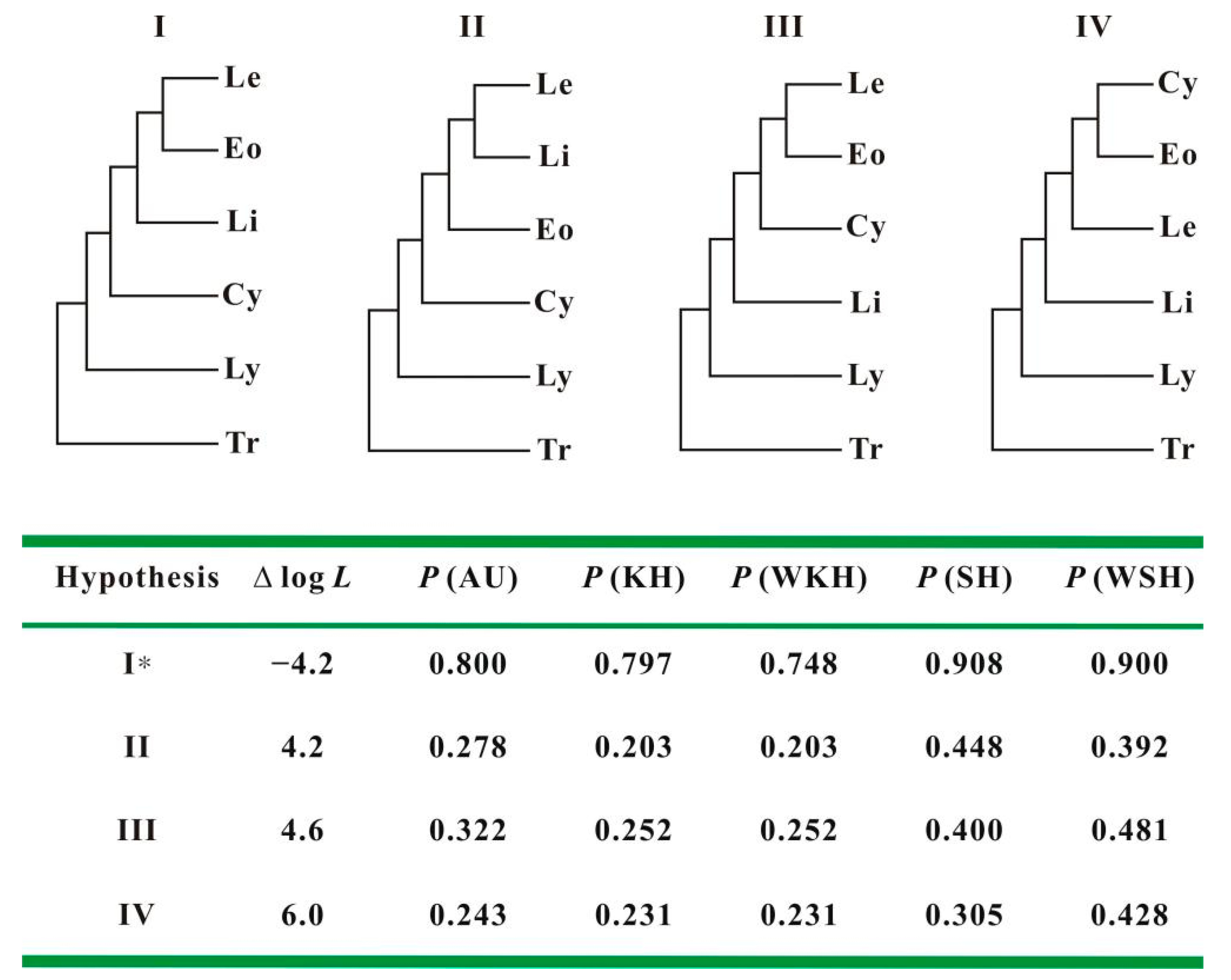
3.4. Testing Support for Competing Hypotheses
4. Discussion
4.1. Ribosomal DNA Polymorphism and Pseudogenes
4.2. 28S rDNA Polymorphism and Mode of Reproduction
4.3. Potential Phylogenetic Signal for Resolving Spinicaudatan Phylogeny
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brendonck, L.; Rogers, D.C.; Olesen, J.; Weeks, S.; Hoeh, W.R. Global diversity of large branchiopods (Crustacea: Branchiopoda) in freshwater. Hydrobiologia 2008, 595, 167–176. [Google Scholar] [CrossRef]
- Rogers, D.C. Branchiopoda (Anostraca, Notostraca, Laevicaudata, Spinicaudata, Cyclestherida). In Encyclopedia of Inland Waters; Likens, G.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 2, pp. 242–249. [Google Scholar]
- Rogers, D.C. Spinicaudata catalogus (Crustacea: Branchiopoda). Zool. Stud. 2020, 59, 45. [Google Scholar] [CrossRef]
- Schwentner, M.; Rabet, N.; Richter, S.; Giribet, G.; Padhye, S.; Cart, J.-F.; Bonillo, C.; Rogers, D.C. Phylogeny and biogeography of Spinicaudata (Crustacea: Branchiopoda). Zool. Stud. 2020, 59, 44. [Google Scholar] [CrossRef]
- Tasch, P. Branchiopoda. In Treatise on Invertebrate Paleontology, Part R, Arthropoda 4; Moore, R.C., Ed.; The University of Kansas and Geological Society of America: Lawrence, KS, USA, 1969; Volume 1, pp. 128–191. [Google Scholar]
- Zhang, W.; Chen, P.; Shen, Y. Fossil Conchostracans of China; Science Press: Beijing, China, 1976. (In Chinese) [Google Scholar]
- Chen, P.; Shen, Y. An Introduction to Fossil Conchostracans; Science Press: Beijing, China, 1985. (In Chinese) [Google Scholar]
- Negrea, S.; Botnariuc, N.; Dumont, H.J. Phylogeny, evolution and classification of the Branchiopoda (Crustacea). Hydrobiologia 1999, 412, 191–212. [Google Scholar] [CrossRef]
- Olesen, J. Monophyly and phylogeny of Branchiopoda, with focus on morphology and homologies of branchiopod phyllopodus limbs. J. Crustac. Biol. 2007, 27, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Richter, S.; Olesen, J.; Wheeler, W.C. Phylogeny of Branchiopoda (Crustacea) based on a combined analysis of morphological data and six molecular loci. Cladistics 2007, 23, 301–336. [Google Scholar] [CrossRef]
- Schwentner, M.; Timms, B.V.; Bastrop, R.; Richter, S. Phylogeny of Spinicaudata (Branchiopoda, Crustacea) based on three molecular markers—An Australian origin for Limnadopsis. Mol. Phylogenet. Evol. 2009, 53, 716–725. [Google Scholar] [CrossRef]
- Hoeh, W.R.; Smallwood, N.D.; Senyo, D.M.; Chapman, E.G.; Weeks, S.C. Evaluating the monophyly of Eulimnadia and the Limnadiinae (Branchiopoda: Spinicaudata) using DNA sequences. J. Crustac. Biol. 2006, 26, 182–192. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J. A phylogenetic analysis of the Conchostraca and Cladocera (Crustacea, Branchiopoda, Diplostraca). Zool. J. Linn. Soc.-Lond. 1998, 122, 491–536. [Google Scholar] [CrossRef]
- Schwentner, M.; Richter, S.; Rogers, D.C.; Giribet, G. Tetraconatan phylogeny with special focus on Malacostraca and Branchiopoda: Highlighting the strength of taxon-specific matrices in phylogenomics. Proc. R. Soc. Lond. B Biol. Sci. 2018, 285, 20181524. [Google Scholar] [CrossRef] [Green Version]
- Weeks, S.C.; Chapman, E.G.; Rogers, D.C.; Senyo, D.M.; Hoeh, W.R. Evolutionary transitions among dioecy, androdioecy and hermaphroditism in limnadiid clam shrimp (Branchiopoda: Spinicaudata). J. Evol. Biol. 2009, 22, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Bellec, L.; Rabet, N. Dating of the Limnadiidae family suggests an American origin of Eulimnadia. Hydrobiologia 2016, 773, 149–161. [Google Scholar] [CrossRef]
- Philippe, H.; Delsuc, F.; Brinkmann, H.; Lartillot, N. Phylogenomics. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 541–562. [Google Scholar] [CrossRef]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesnidal, M.P.; Helmkampf, M.; Bruchhaus, I.; Hausdorf, B. Compositional heterogeneity and phylogenomic inference of metazoan relationships. Mol. Biol. Evol. 2010, 27, 2095–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dover, G. Molecular drive: A cohesive mode of species evolution. Nature 1982, 299, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Crease, T.; Lynch, M. Ribosomal DNA variation in Daphnia pulex. Mol. Biol. Evol. 1991, 8, 620–640. [Google Scholar] [CrossRef] [Green Version]
- Hugall, A.; Stanton, J.; Moritz, C. Reticulate evolution and the origins of ribosomal internal transcribed spacer diversity in apomictic Meloidogyne. Mol. Biol. Evol. 1999, 16, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Bik, H.M.; Fournier, D.; Sung, W.; Bergeron, R.D.; Thomas, W.K. Intra-genomic variation in the ribosomal repeats of nematodes. PLoS ONE 2013, 8, e78230. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Rooney, A.P. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 2005, 39, 121–152. [Google Scholar] [CrossRef] [Green Version]
- Higgs, P. RNA secondary structure: Physical and computational aspects. Q. Rev. Biophys. 2000, 33, 199–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letsch, H.O.; Kjer, K.M. Potential pitfalls of moddelling ribosomal RNA data in phylogenetic tree reconstruction: Evidence from case studies in the Metazoa. BMC Evol. Biol. 2011, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.; Steel, M. How much can evolved characters tell us about the tree that generated them? In Mathematics of Evolution and Phylogeny; Gascuel, O., Ed.; Oxford University Press: Oxford, UK, 2005; pp. 384–412. [Google Scholar]
- Xiong, B.; Kocher, T.D. Comparison of mitochondrial DNA sequences of seven morphospecies of balck flies (Diptera: Simuliidae). Genome 1991, 34, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Sonnenberg, R.; Nolte, A.W.; Tautz, D. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification. Front. Zool. 2007, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, T.D.; Taylor, D.J. Structural rRNA characters support monophyly of raptorial limbs and paraphyly of limb specialization in water fleas. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crease, T.J. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene 1999, 233, 89–99. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, J. Characterization of the complete mitochondrial genome of Chinese Triops granarius and implications for species delimitation. Int. J. Biol. Macromol. 2019, 135, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNA structure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Schöniger, M.; von Haeseler, A. A stochastic model for the evolution of autocorrelated DNA sequences. Mol. Phylogenet. Evol. 1994, 3, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Huelsenbeck, J.P. MrBayes 3.2: Effificient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylander, J.A.A. MrModeltest Version 2; Program Distributed by the Author; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004; Available online: https://github.com/nylander/MrModeltest2 (accessed on 25 October 2021).
- Brandley, M.C.; Schmitz, A.; Reeder, T.W. Partitioned Bayesian analyses, partition choice, and the phylogenetic relationships of scincid lizards. Syst. Biol. 2005, 54, 373–390. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Shimodaira, H. An approximately unbiased test of phylogenetic treeselection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [Green Version]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of loglikelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Shimodaira, H.; Hasegawa, M. CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics 2001, 17, 1246–1247. [Google Scholar] [CrossRef] [Green Version]
- Schnare, M.N.; Damberger, S.H.; Gray, M.W.; Gutell, R.R. Comprehensive comparison of structural characteristics in eukaryotic cytoplasmic large subunit (23 S-like) ribosomal RNA. J. Mol. Biol. 1996, 256, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.J.; Cannone, J.J.; Gutell, R.R.; Cognato, A.I. A secondary structural model of the 28S rRNA expansion segments D2 and D3 from rootworms and related leaf beetles (Coleoptera: Chrysomelidae; Galerucinae). Insect Mol. Biol. 2004, 13, 495–518. [Google Scholar] [CrossRef]
- Petrov, A.S.; Bernier, C.R.; Gulen, B.; Waterbury, C.C.; Hershkovits, E.; Hsiao, C.; Harvey, S.C.; Hud, N.V.; Fox, G.E.; Wartell, R.M.; et al. Secondary structures of rRNAs from all three domains of life. PLoS ONE 2014, 9, e88222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutell, R.R.; Cannone, J.J.; Shang, Z.; Du, Y.; Serra, M.J. A story: Unpaired adenosine bases in ribosomal RNAs. J. Mol. Biol. 2000, 304, 335–354. [Google Scholar] [CrossRef]
- Schwentner, M.; Just, F.; Richter, S. Evolutionary systematics of the Australian Cyzicidae (Crustacea, Branchiopoda, Spinicaudata) with the description of a new genus. Zool. J. Linn. Soc.-Lond. 2015, 173, 271–295. [Google Scholar] [CrossRef] [Green Version]
- Brantner, J.S.; Ott, D.W.; Duff, R.J.; Orridge, J.I.; Waldman, J.R.; Weeks, S.C. Evidence of selfing hermaphroditism in the clam shrimp Cyzicus gynecia (Branchiopoda: Spinicaudata). J. Crustac. Biol. 2013, 33, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Brantner, J.S.; Ott, D.W.; Duff, R.J.; Sanoamuang, L.; Simhachalam, G.P.; Babu, K.K.S.; Weeks, S.C. Androdioecy and hermaphroditism in five species of clam shrimp (Crustacea: Branchiopoda: Spinicaudata) from India and Thailand. Invertebr. Biol. 2013, 132, 27–37. [Google Scholar] [CrossRef]
- Sigvardt, Z.M.S.; Rogers, D.C.; Olesen, J. Functional morphology of amplexus (clasping) in spinicaudatan clam shrimps (Crustacea, Branchiopoda) and its evolution in bivalved branchiopods: A video-based analysis. J. Morphol. 2017, 278, 523–546. [Google Scholar] [CrossRef] [PubMed]
- Weeks, S.C.; Benvenuto, C.; Reed, S.K. When males and hermaphrodites coexist: A review of androdioecy in animals. Integr. Comp. Biol. 2006, 46, 449–464. [Google Scholar] [CrossRef]
- Weeks, S.C.; Sanderson, T.F.; Reed, S.K.; Zofkova, M.; Knott, B.; Balaraman, U.; Pereira, G.; Senyo, D.M.; Hoeh, W.R. Ancient androdioecy in the freshwater crustacean Eulimnadia. Proc. R. Soc. Lond. B Biol. Sci. 2006, 273, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, S.C.; Sanderson, T.F.; Zofkova, M.; Knott, B. Breeding systems in the clam shrimp family Limnadiidae (Branchiopoda, Spinicaudata). Invertebr. Biol. 2008, 127, 336–349. [Google Scholar] [CrossRef]
- Weeks, S.C.; Benvenuto, C.; Sanderson, T.F.; Duff, R.J. Sex chromosome evolution in the clam shrimp, Eulimnadia texana. J. Evol. Biol. 2010, 23, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Weeks, S.C.; Brantner, J.S.; Astrop, T.I.; Ott, D.W.; Rabet, N. The evolution of hermaphroditism from dioecy in crustaceans: Selfing hermaphroditism described in a fourth spinicaudatan genus. Evol. Biol. 2014, 41, 251–261. [Google Scholar] [CrossRef]
- Hartmann, S.; Vision, T.J. Using ESTs for phylogenomics: Can one accurately infer a phylogenetic tree from a gappy alignment? BMC Evol. Biol. 2008, 8, 95. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Driver, F.; Barro, P.D. The phylogenetic characteristics of three different 28S rRNA gene regions in Encarsia (Insecta, Hymenoptera, Aphelinidae). Org. Divers. Evol. 2006, 6, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Shi, W.; Yang, M.; Luo, H. Variations in the conserved 18S and 5.8S reveal the putative pseudogenes in 18S-ITS1-5.8S rDNA of Cynoglossus melampetalus (Pleuronectiformes: Cynoglossidae). Biochem. Biophys. Res. Commun. 2021, 534, 233–239. [Google Scholar] [CrossRef]
- Smith, S.D.; Bond, J.E. An analysis of the secondary structure of the mitochondrial large subunit rRNA gene (16S) in spiders and its implications for phylogenetic reconstruction. J. Arachnol. 2003, 31, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Buckley, T.R.; Simon, C.; Flook, P.K.; Misof, B. Secondary structure and conserved motifs of the frequently sequenced domains iv and v of the insect mitochondrial large subunit rRNA gene. Insect Mol. Biol. 2000, 9, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, J.J.; Yoder, M.J.; Wharton, R.A. Predicted Secondary Structure for 28S and 18S rRNA from Ichneumonoidea (Insecta: Hymenoptera: Apocrita): Impact on Sequence Alignment and Phylogeny Estimation. J. Mol. Evol. 2005, 61, 114–137. [Google Scholar] [CrossRef] [PubMed]
- Sassaman, C. Sex determination and evolution of unisexuality in the Conchostraca. Hydrobiologia 1995, 298, 45–65. [Google Scholar] [CrossRef]
- Weeks, S.C. The role of androdioecy and gynodioecy in mediating evolutionary transitions between dioecy and hermaphroditism in the Animalia. Evolution 2012, 66, 3670–3686. [Google Scholar] [CrossRef] [PubMed]
- Zierold, T.; Montero-Pau, J.; Hanfling, B.; Gómez, A. Sex ratio, reproductive mode and genetic diversity in Triops cancriformis. Freshw. Biol. 2009, 54, 1392–1405. [Google Scholar] [CrossRef]
- Márquez, L.M.; Miller, D.J.; MacKenzie, J.B.; van Oppen, M.J.H. Pseudogenes contribute to the extreme diversity of nuclear ribosomal DNA in the hard coral Acropora. Mol. Biol. Evol. 2003, 20, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brendonck, L.; Thiery, A.; Coomans, A. Taxonomy and biogeography of the Galapagos branchiopod fauna (Anostraca, Notostraca, Spinicaudata). J. Crustac. Biol. 1990, 10, 676–694. [Google Scholar] [CrossRef]
- Bellec, L.; Debruyne, R.; Utge, J.; Rabet, N. The first complete mitochondrial genome of Limnadia lenticularis (Branchiopoda, Spinicaudata), with new insights on its phylogeography and on the taxonomy of the genus. Hydrobiologia 2019, 826, 145–158. [Google Scholar] [CrossRef]
- Springer, M.S.; DeBry, R.W.; Douady, C.; Amrine, H.M.; Madsen, O.; de Jong, W.W.; Stanhope, M.J. Mitochondrial versus nuclear gene sequences in deep-level mammalian phylogeny reconstruction. Mol. Biol. Evol. 2001, 18, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Rokas, A.; Carroll, S.B. Frequent and widespread parallel evolution of protein sequences. Mol. Biol. Evol. 2008, 25, 1943–1953. [Google Scholar] [CrossRef] [Green Version]
- Townsend, J.P.; Su, Z.; Tekle, Y.I. Phylogenetic Signal and Noise: Predicting the Power of a Data Set to Resolve Phylogeny. Syst. Biol. 2012, 61, 835–849. [Google Scholar] [CrossRef]
- Feuda, R.; Smith, A.B. Phylogenetic signal dissection identifies the root of starfishes. PLoS ONE 2015, 10, e0123331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, D. Identifying and removing fast-evolving sites using compatibility analysis: An example from the Arthropoda. Syst. Biol. 2004, 53, 978–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, D.; Feuda, R.; Peterson, K.J.; Smith, A.B. Resolving phylogenetic signal from noise when divergence is rapid: A new look at the old problem of echinoderm class relationships. Mol. Phylogenet. Evol. 2012, 62, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Rota-Stabelli, O.; Campbell, L.; Brinkmann, H.; Edgecombe, G.D.; Longhorn, S.J.; Peterson, K.J. A congruent solution to arthropod phylogeny: Phylogenomics, microRNAs and morphology support monophyletic Mandibulata. Proc. R. Soc. Lond. B Biol. Sci. 2011, 278, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, E.A.; Pisani, D.; Peterson, K.J. Molecular paleobiological insights into the origin of the Brachiopoda. Evol. Dev. 2011, 13, 290–303. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Gene | Species | Length * | P | Iss | Iss.c | P | C | V | Pi | S |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyzicus | 28S | 7 | 537 | stem | 0.411 | 0.777 | 0.00 | 333 | 27 | 11 | 16 |
| loop | 1.915 | 0.679 | 0.00 | 149 | 14 | 7 | 7 | ||||
| 16S | 9 | 545 | stem | 2.133 | 0.644 | 0.00 | 235 | 37 | 34 | 3 | |
| loop | 1.370 | 0.645 | 0.00 | 198 | 58 | 50 | 8 | ||||
| Ozestheria | 28S | 27 | 549 | stem | 1.074 | 0.778 | 0.00 | 322 | 48 | 36 | 12 |
| loop | 1.021 | 0.595 | 0.04 | 144 | 26 | 9 | 17 | ||||
| 16S | 24 | 570 | stem | 0.643 | 0.659 | 0.88 | 214 | 67 | 53 | 14 | |
| loop | 1.254 | 0.682 | 0.00 | 172 | 96 | 71 | 25 | ||||
| Eocyzicus | 28S | 12 | 531 | stem | 1.043 | 0.777 | 0.00 | 322 | 32 | 6 | 25 |
| loop | 1.470 | 0.651 | 0.00 | 156 | 14 | 4 | 10 | ||||
| 16S | 20 | 517 | stem | 0.634 | 0.644 | 0.92 | 234 | 35 | 28 | 7 | |
| loop | 0.730 | 0.631 | 0.38 | 191 | 56 | 40 | 16 | ||||
| Leptestheria +Eoleptestheria +Maghrebestheria | 28S | 12 | 523 | stem | 0.170 | 0.777 | 0.00 | 327 | 26 | 22 | 4 |
| loop | 0.922 | 0.664 | 0.16 | 153 | 17 | 13 | 4 | ||||
| 16S | 16 | 517 | stem | 0.484 | 0.657 | 0.05 | 190 | 81 | 41 | 40 | |
| loop | 0.930 | 0.665 | 0.04 | 162 | 56 | 40 | 16 | ||||
| Paralimnadia | 28S | 7 | 477 | stem | 0.089 | 0.783 | 0.00 | 291 | 23 | 6 | 17 |
| loop | 1.869 | 0.603 | 0.00 | 145 | 10 | 4 | 6 | ||||
| 16S | 2 | 460 | stem | - | - | - | - | - | - | - | |
| loop | - | - | - | - | - | - | - | ||||
| Limnadopsis | 28S | 6 | 469 | stem | 0.015 | 0.780 | 0.00 | 300 | 12 | 11 | 1 |
| loop | 0.206 | 0.647 | 0.00 | 151 | 9 | 6 | 3 | ||||
| 16S | 4 | 515 | stem | 0.328 | 0.779 | 0.00 | 209 | 12 | 3 | 9 | |
| loop | 1.445 | 0.777 | 0.00 | 185 | 20 | 4 | 16 | ||||
| Eulimnadia | 28S | 16 | 482 | stem | 0.891 | 0.773 | 0.50 | 281 | 34 | 24 | 10 |
| loop | 2.401 | 0.585 | 0.00 | 148 | 17 | 15 | 2 | ||||
| 16S | 4 | 530 | stem | 0.251 | 0.780 | 0.00 | 263 | 12 | 2 | 10 | |
| loop | 1.723 | 0.758 | 0.00 | 227 | 10 | 3 | 7 | ||||
| Spinicaudata | 28S & 16S & EF1α & COI | 40 | 2092 | Stem, loop, 1st&2nd | 0.614 | 0.807 | 0.00 | 1202 | 845 | 652 | 186 |
| Item | Hypothesis | ∆ log L | p Values | ||||
|---|---|---|---|---|---|---|---|
| AU | KH | WKH | SH | WSH | |||
| 1 | ((((Limnadopsis, Paralimnadia), Australimnadia), Eulimnadia), (Imnadia, Limnadia)) | −1.5 | 0.676 | 0.651 | 0.651 | 0.818 | 0.835 |
| 2 | (((((Limnadopsis, Paralimnadia), Australimnadia), Eulimnadia), Imnadia), Limnadia) | 1.5 | 0.436 | 0.349 | 0.349 | 0.691 | 0.667 |
| 4 | (((((Limnadopsis, Paralimnadia), Australimnadia), Limnadia), Imnadia), Eulimnadia) | 3.6 | 0.335 | 0.317 | 0.317 | 0.457 | 0.570 |
| 3 | ((((Limnadopsis, Paralimnadia), Australimnadia), (Imnadia, Eulimnadia)), Limnadia) | 10.5 | 0.018 | 0.044 | 0.044 | 0.087 | 0.109 |
| Item | Hypothesis * | ∆ log L | p Values | ||||
|---|---|---|---|---|---|---|---|
| AU | KH | WKH | SH | WSH | |||
| 1 | (((((Eole, Magh), Lkaw), ((Ldah, Lcor), Lrub), ((Lbre, Lspm), Lnob)), Lven), Lcom) | −628.1 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| 2 | ((((((((Ldah, Lcor), Lrub), ((Lbre, Lspm), Lnob)), Lven), Lcom), (Eole, Lspm)), Lkaw), Magh) | 628.1 | 6 × 10−6 ** | 0.000 | 0.000 | 0.000 | 0.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Cheng, J. Phylogenetic Signal Dissection of Heterogeneous 28S and 16S rRNA Genes in Spinicaudata (Branchiopoda, Diplostraca). Genes 2021, 12, 1705. https://doi.org/10.3390/genes12111705
Sun X, Cheng J. Phylogenetic Signal Dissection of Heterogeneous 28S and 16S rRNA Genes in Spinicaudata (Branchiopoda, Diplostraca). Genes. 2021; 12(11):1705. https://doi.org/10.3390/genes12111705
Chicago/Turabian StyleSun, Xiaoyan, and Jinhui Cheng. 2021. "Phylogenetic Signal Dissection of Heterogeneous 28S and 16S rRNA Genes in Spinicaudata (Branchiopoda, Diplostraca)" Genes 12, no. 11: 1705. https://doi.org/10.3390/genes12111705
APA StyleSun, X., & Cheng, J. (2021). Phylogenetic Signal Dissection of Heterogeneous 28S and 16S rRNA Genes in Spinicaudata (Branchiopoda, Diplostraca). Genes, 12(11), 1705. https://doi.org/10.3390/genes12111705