
Genome-Wide Landscape of North-Eastern European Populations: A View from Lithuania




Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Genotyping
2.3. Admixture and Principal Component Analysis
2.4. Ne and Divergence Time Analysis
2.5. Selection Signatures
3. Results
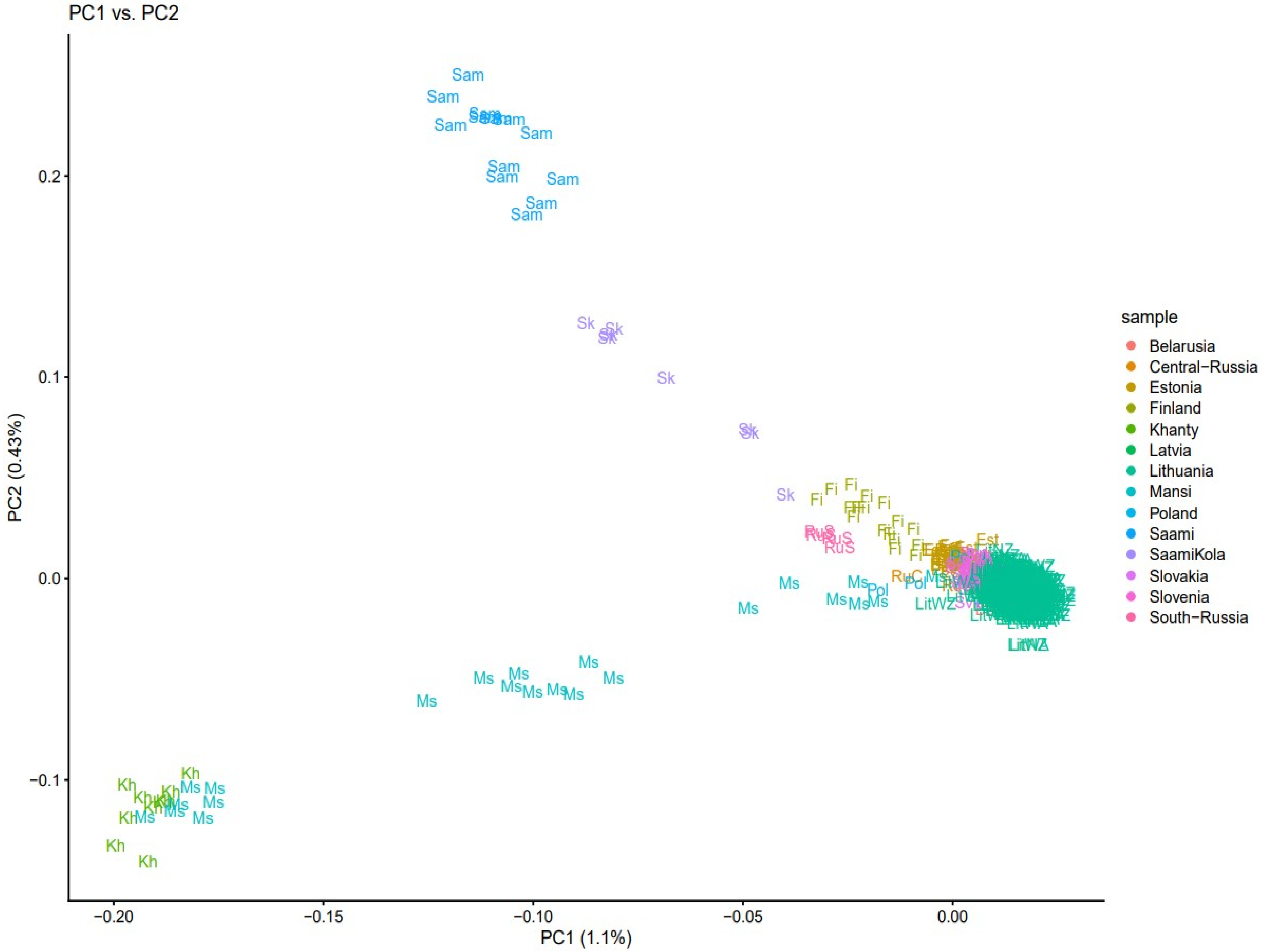
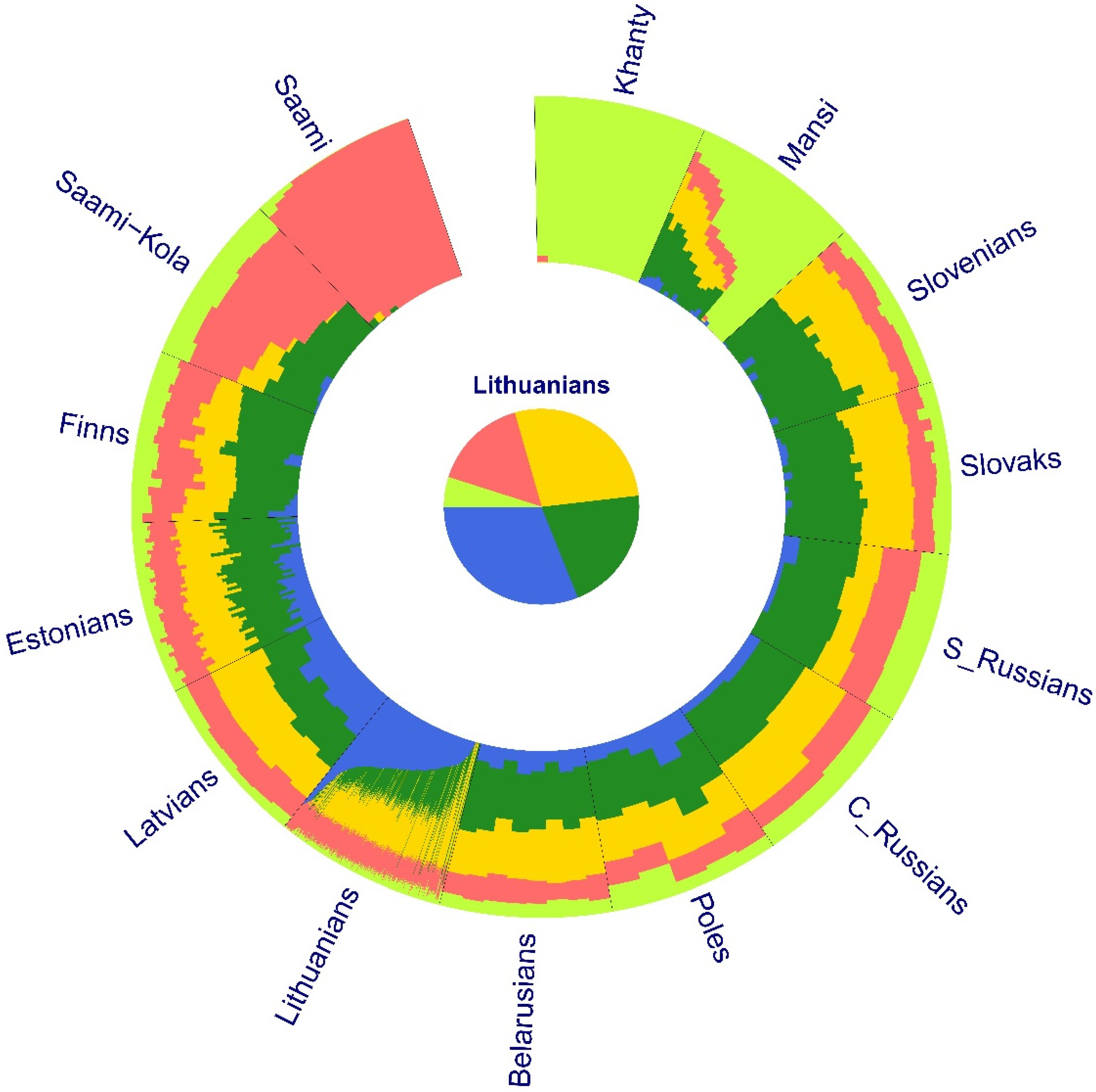
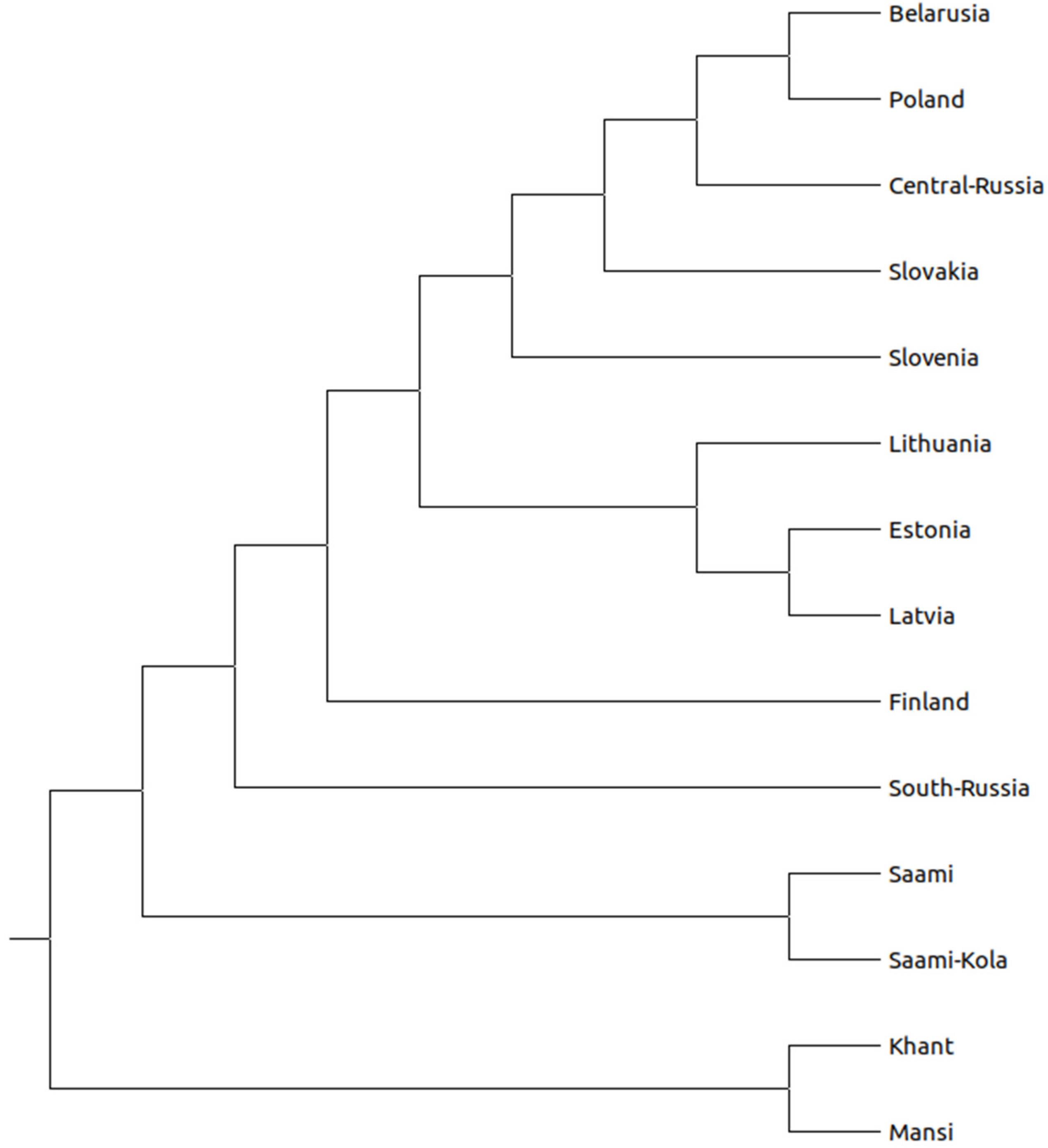
3.1. Population Structure and Divergence Time Analysis
3.2. Identifying Regions under Recent Positive Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Urnikyte, A.; Flores-Bello, A.; Mondal, M.; Molyte, A.; Comas, D.; Calafell, F.; Bosch, E.; Kučinskas, V. Patterns of genetic structure and adaptive positive selection in the Lithuanian population from high-density SNP data. Sci. Rep. 2019, 9, 9163. [Google Scholar] [CrossRef] [PubMed]
- Klimas, A. Some unique futures of Lithuanian. Lituanus 1984, 30, 51–64. [Google Scholar]
- Laitinen, V.; Lahermo, P.; Sistonen, P.; Savontaus, M.-L. Y-Chromosomal Diversity Suggests that Baltic Males Share Common Finno-Ugric-Speaking Forefathers. Hum. Hered. 2002, 53, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Rimantienė, R. Akmens Amžius Lietuvoje; Žiburys: Vilnius, Lithuania, 1966. [Google Scholar]
- Zilinskas, J. Lietuvių Protėviai (The Ancestors of Lithuanians); Raides: Kaunas, Lithuania, 1937; pp. 1–143. [Google Scholar]
- Gimbutas, M. The Indo-Europeans: Archeological Problems. Am. Anthropol. 1963, 65, 815–836. [Google Scholar] [CrossRef]
- Cesnys, G.; Kucinskas, V. The Balts and the Finns in historical perspective: A multidisciplinary approach. Acta Med. Litu. 2004, 11, 7. [Google Scholar]
- Biddiss, M.D. The Europeans—An Ethnohistorical Survey. By John Geipel (London, Longmans, Green and Company Limited, 1969). xii + 292 pp 60s. Race 1970, 11, 387–388. [Google Scholar] [CrossRef]
- Kasperavičiūtė, D.; Kučinskas, V.; Stoneking, M. Y Chromosome and Mitochondrial DNA Variation in Lithuanians: Y Chomosome and mtDNA Variation in Lithuanians. Ann. Hum. Genet. 2004, 68, 438–452. [Google Scholar] [CrossRef] [Green Version]
- Harvey, R.G.; Tills, D.; Warlow, A.; Kopec, A.C.; Domaniewska-Sobczak, K.; Suter, D.; Lord, J. Genetic Affinities of the Balts: A Study of Blood Groups, Serum Proteins and Enzymes of Lithuanians in the United Kingdom. Man 1983, 18, 535–552. [Google Scholar] [CrossRef]
- Pankratov, V.; Montinaro, F.; Kushniarevich, A.; Hudjashov, G.; Jay, F.; Saag, L.; Flores, R.; Marnetto, D.; Seppel, M.; Kals, M.; et al. Differences in local population history at the finest level: The case of the Estonian population. Eur. J. Hum. Genet. 2020, 28, 1580–1591. [Google Scholar] [CrossRef]
- Tambets, K.; Yunusbayev, B.; Hudjashov, G.; Ilumäe, A.-M.; Rootsi, S.; Honkola, T.; Vesakoski, O.; Atkinson, Q.; Skoglund, P.; Kushniarevich, A.; et al. Genes reveal traces of common recent demographic history for most of the Uralic-speaking populations. Genome Biol. 2018, 19, 139. [Google Scholar] [CrossRef] [Green Version]
- Kushniarevich, A.; Utevska, O.; Chuhryaeva, M.; Agdzhoyan, A.; Dibirova, K.; Uktveryte, I.; Möls, M.; Mulahasanovic, L.; Pshenichnov, A.; Frolova, S.; et al. Genetic Heritage of the Balto-Slavic Speaking Populations: A Synthesis of Autosomal, Mitochondrial and Y-Chromosomal Data. PLoS ONE 2015, 10, e0135820. [Google Scholar] [CrossRef] [Green Version]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2007, 6, 2867–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, A.A.; Liu, K.Z.; Liu-Fang, G.; Nakka, P.; Ramachandran, S. Pong: Fast analysis and visualization of latent clusters in population genetic data. Bioinformatics 2016, 32, 2817–2823. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Lu, D.; Xu, S. AncestryPainter: A Graphic Program for Displaying Ancestry Composition of Populations and Individuals. Genom. Proteom. Bioinform. 2018, 16, 382–385. [Google Scholar] [CrossRef]
- Mezzavilla, M. Neon: An R Package to Estimate Human Effective Population Size and Divergence Time from Patterns of Linkage Disequilibrium between SNPS. J. Comput. Sci. Syst. Biol. 2015, 8, 37–44. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef] [PubMed]
- Benazzo, A.; Panziera, A.; Bertorelle, G. 4P: Fast computing of population genetics statistics from large DNA polymorphism panels. Ecol. Evol. 2015, 5, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- The International HapMap Consortium; Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Delaneau, O.; Zagury, J.-F. Haplotype Inference. In Data Production and Analysis in Population Genomics; Pompanon, F., Bonin, A., Eds.; Humana Press: Totowa, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deming, Y.; Filipello, F.; Cignarella, F.; Cantoni, C.; Hsu, S.; Mikesell, R.; Li, Z.; Del-Aguila, J.L.; Dube, U.; Farias, F.G.; et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 2019, 11, eaau2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease (GERAD); Alzheimer’s Disease Genetic Consortium (ADGC); Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE); Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Gen. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- The International Genomics of Alzheimer’s Project; The Alzheimer’s Disease Neuroimaging Initiative; Huang, K.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesanto, A.; Crocco, P.; Tallaro, F.; Pisani, F.; Mazzei, B.; Mari, V.; Corsonello, A.; Lattanzio, F.; Passarino, G.; Rose, G. Common polymorphisms in nitric oxide synthase (NOS) genes influence quality of aging and longevity in humans. Biogerontology 2013, 14, 177–186. [Google Scholar] [CrossRef]
- Pullat, J.; Fleischer, R.; Becker, N.; Beier, M.; Metspalu, A.; Hoheisel, J.D. Optimization of candidate-gene SNP-genotyping by flexible oligonucleotide microarrays; analyzing variations in immune regulator genes of hay-fever samples. BMC Genom. 2007, 8, 282. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.H.M.; Khrunin, A.; Nichols, L.; Pushkarev, D.; Khokhrin, D.; Verbenko, D.; Evgrafov, O.; Knowles, J.; Novembre, J.; Limborska, S.; et al. Reconstructing genetic history of Siberian and Northeastern European populations. Genome Res. 2017, 27, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Urnikytė, A.; Molytė, A.; Pranckevičienė, E.; Kučinskienė, Z.A.; Kučinskas, V. Inferring Effective Population Size and Divergence Time in the Lithuanian Population According to High-Density Genotyping Data. Genes 2020, 11, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shnayder, N.A.; Petrova, M.M.; Moskaleva, P.V.; Shesternya, P.A.; Pozhilenkova, E.A.; Nasyrova, R.F. The Role of Single-Nucleotide Variants of NOS1, NOS2, and NOS3 Genes in the Comorbidity of Arterial Hypertension and Tension-Type Headache. Molecules 2021, 26, 1556. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, F.M.; Luizon, M.R.; Speciali, J.G.; Martins-Oliveira, A.; Dach, F.; Tanus-Santos, J.E. Interaction among nitric oxide (NO)-related genes in migraine susceptibility. Mol. Cell Biochem. 2012, 370, 183–189. [Google Scholar] [CrossRef]
- Iacopetta, B.; Grieu, F.; Amanuel, B. Microsatellite instability in colorectal cancer: MSI and colorectal cancer. Asia Pac. J. Clin. Oncol. 2010, 6, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, D.; Saito, M.; Saito, K.; Watanabe, Y.; Matsumoto, Y.; Kanke, Y.; Onozawa, H.; Hayase, S.; Sakamoto, W.; Ishigame, T.; et al. Upregulated solute carrier family 37 member 1 in colorectal cancer is associated with poor patient outcome and metastasis. Oncol. Lett. 2017, 15, 2065–2072. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Genes | Population (SNPs *) |
|---|---|---|
| chr1:80069451-80662898 | ADGRL4, LINC01781 | LT-LVL(2) |
| chr1:245924864-246512218 | SMYD3 | LT-Khanty(3) LT-Mansi(12) |
| chr2:159061258-159558658 | CCDC148-AS1, CCDC148, PKP4, PKP4-AS1 | LT-LVL(7) |
| chr2:153248404-154751502 | FMNL2, RPRM, GALNT13 | LT-SVK(4) |
| chr4:25467149-25705912 | ANAPC4, LOC101929161, LOC101929161, SLC34A2, SEL1L3 | LT-EST(2) |
| chr6:24507761-24575094 | ALDH5A1, KIAA0319 | LT-EST(2) |
| chr6:625268-711792 | EXOC2, LOC101927691 | LT-LVL(3) |
| chr6:109742015-110156179 | PPIL6, AK9, FIG4 | LT-Mansi(4) |
| chr6:28018944-28630691 | OR2B6, OR1F12, ZKSCAN8, ZNF192P1, TOB2P1, ZSCAN9, ZKSCAN4, NKAPL, PGBD1, ZSCAN31, ZSCAN12, ZSCAN23, GPX6, GPX5, ZBED9 | LT-SVK(6) |
| chr7:19566286-20049554 | FERD3L, TWISTNB, TMEM196, LOC101927668 | LT-LVL(2) |
| chr8:60549318-61722552 | LOC100505501, CA8, CHD7 | LT-SaamiKola(8) |
| chr9:126324050-126690157 | DENND1A | LT-POL(4) |
| chr9:12483221-12709305 | PTPRD-AS2, TYRP1, LURAP1L-AS1 | LT-Mansi(2)/ LT-Khanty(6) |
| chr11:60050125-60223018 | MS4A4A, MS4A6E, MS4A7, MS4A14, MS4A1 | LT-LVL(4) |
| chr11:83986071-86064757 | DLG2, PICALM, EED, HIKESHI, CCDC81 | LT-SVK(2) |
| chr12:19199330-19698168 | CAPZA3, PLEKHA5, AEBP2, LINC02398 | LT-Mansi(4) |
| chr12:27569063-28236948 | ARNTL2-AS1, SMCO2, PPFIBP1, KLHL42, PTHLH, LOC729291 | LT-SVK(2) |
| chr12:83895440-84204074 | TMTC2, SLC6A15 | LT-SVK(9) |
| chr14:96157187-96227199 | TCL1B, TCL1A, LOC107984703, TUNAR | LT-SaamiKola(2) |
| chr16:82718030-82822631 | CDH13, LOC101928446 | LT-POL(2) |
| chr20:9325269-9380556 | PLCB4 | LT-BEL(2) |
| chr20:42488256-42593220 | GTSF1L, LINC01728, TOX2 | LT-Mansi(4) |
| chr21:21388493-21644798 | LINC01683, LINC02573 | LT-SaamiKola(6) |
| chr21:43992477-44066201 | SLC37A1, LINC01671, PDE9A | LT-SVK(3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urnikyte, A.; Molyte, A.; Kučinskas, V. Genome-Wide Landscape of North-Eastern European Populations: A View from Lithuania. Genes 2021, 12, 1730. https://doi.org/10.3390/genes12111730
Urnikyte A, Molyte A, Kučinskas V. Genome-Wide Landscape of North-Eastern European Populations: A View from Lithuania. Genes. 2021; 12(11):1730. https://doi.org/10.3390/genes12111730
Chicago/Turabian StyleUrnikyte, Alina, Alma Molyte, and Vaidutis Kučinskas. 2021. "Genome-Wide Landscape of North-Eastern European Populations: A View from Lithuania" Genes 12, no. 11: 1730. https://doi.org/10.3390/genes12111730
APA StyleUrnikyte, A., Molyte, A., & Kučinskas, V. (2021). Genome-Wide Landscape of North-Eastern European Populations: A View from Lithuania. Genes, 12(11), 1730. https://doi.org/10.3390/genes12111730