
Comparative RNA-Seq Analysis Reveals Potentially Resistance-Related Genes in Response to Bacterial Canker of Tomato

 , , and
, , and
Abstract
:
1. Introduction
2. Results
2.1. Infection of Tomato Plants with Cmm 1569
2.2. Mappings Stats and Transcript Quantification
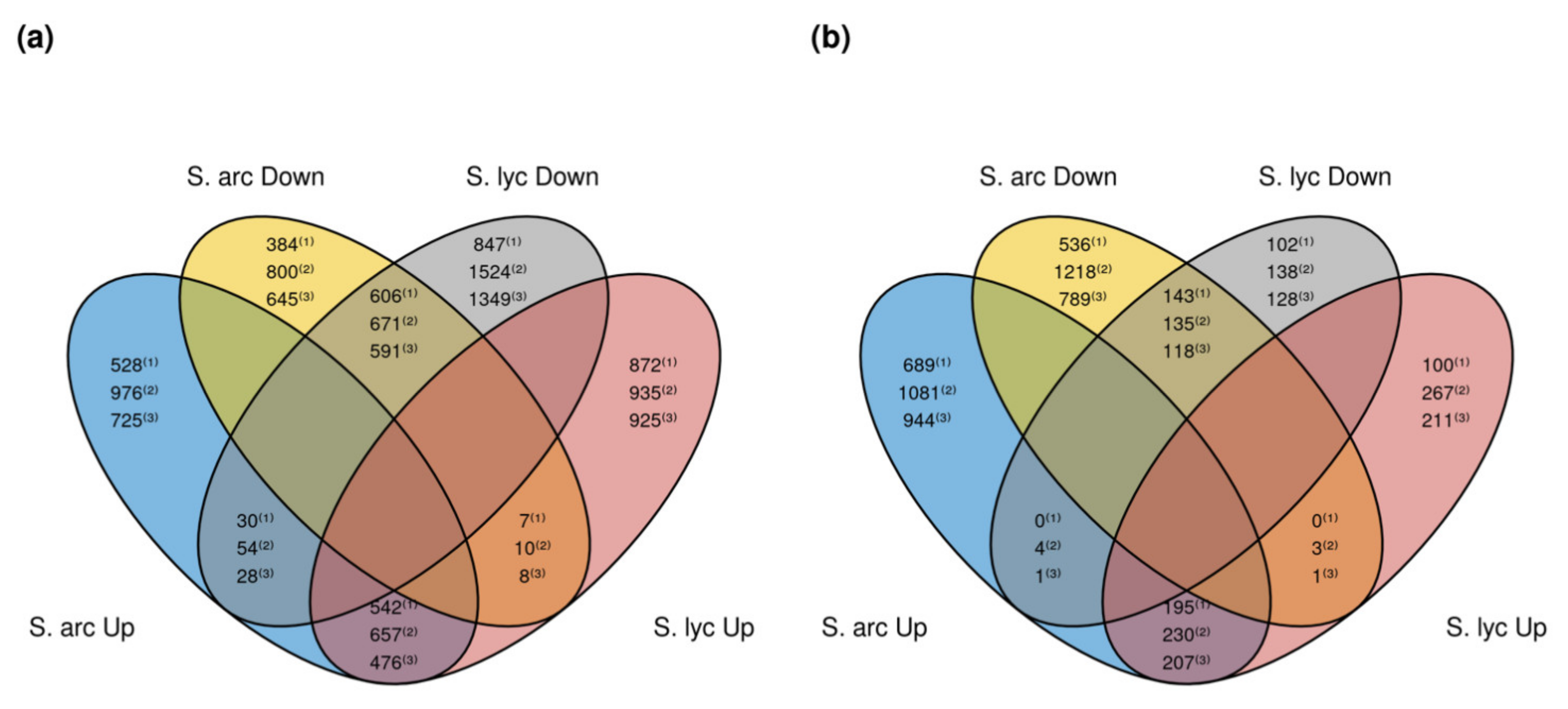
2.3. Differentially Expressed Genes in Response to Cmm 1569
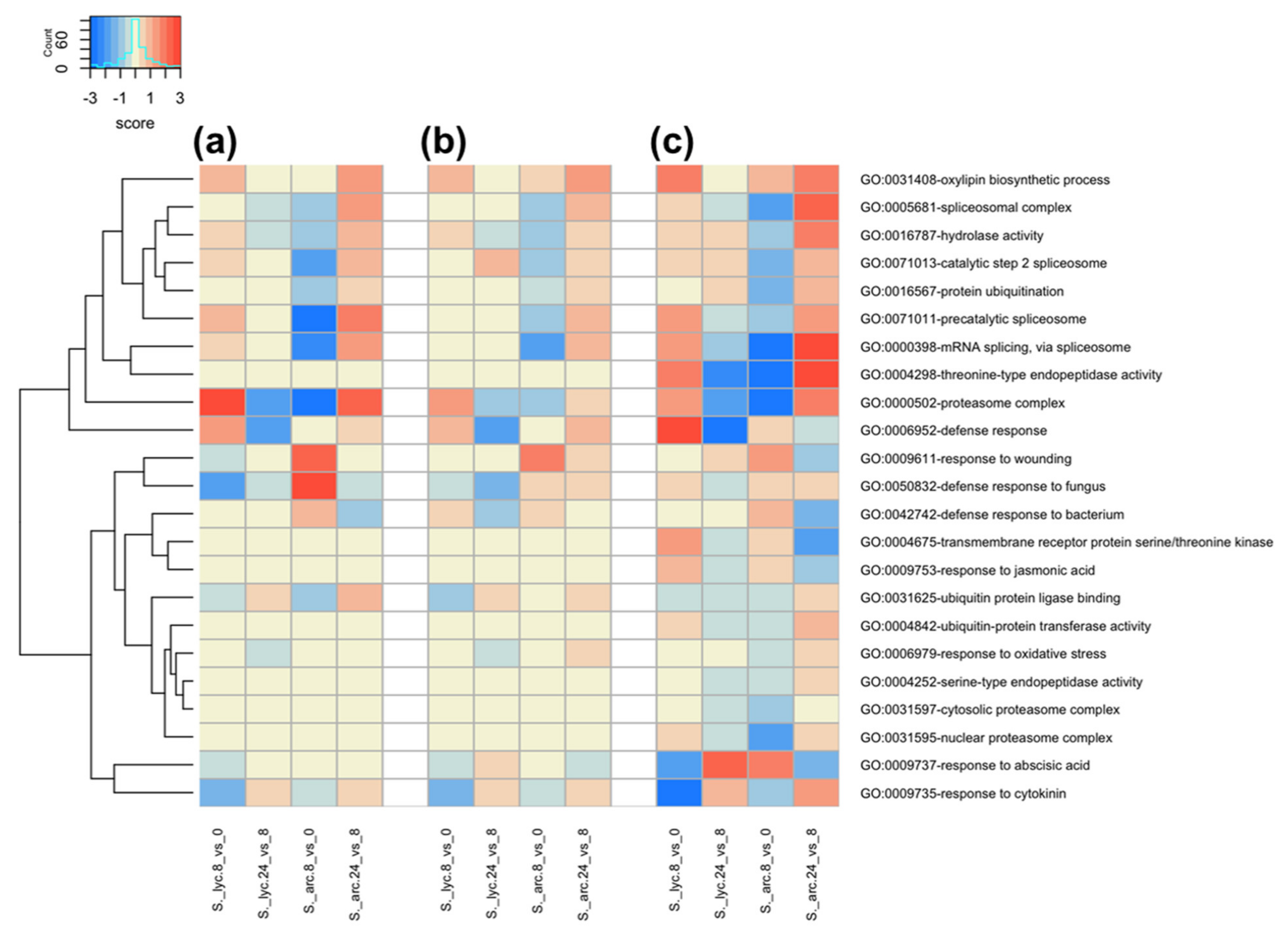
2.4. Gene Ontology Term Enrichment Analysis in S. lycopersicum and S. arcanum LA2157
2.5. DEGs Presumably Associated with the Defense Response against Cmm 1569
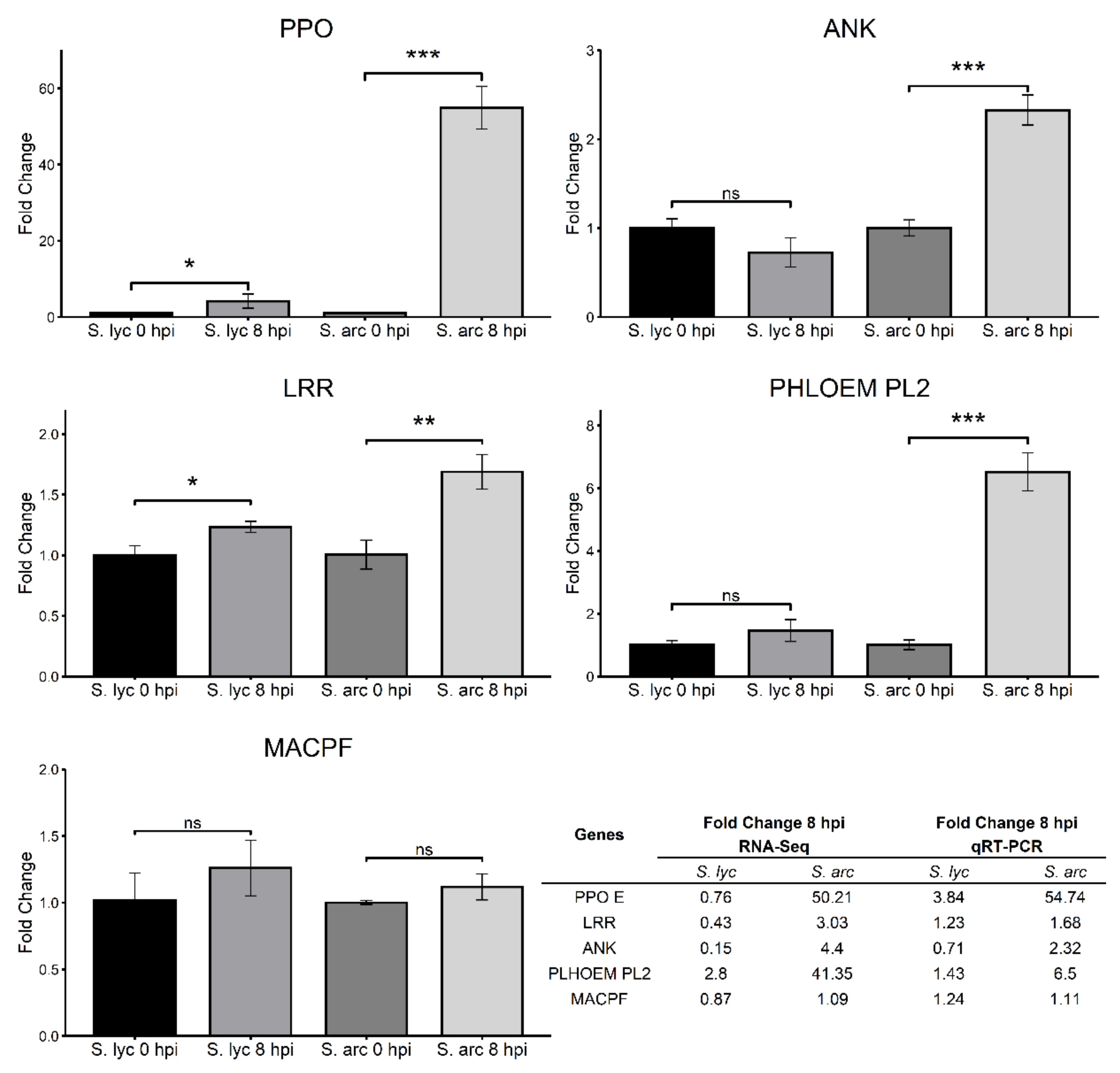
2.6. RNA-Seq Validation Using DEGs Potentially Related to Resistance
3. Discussion
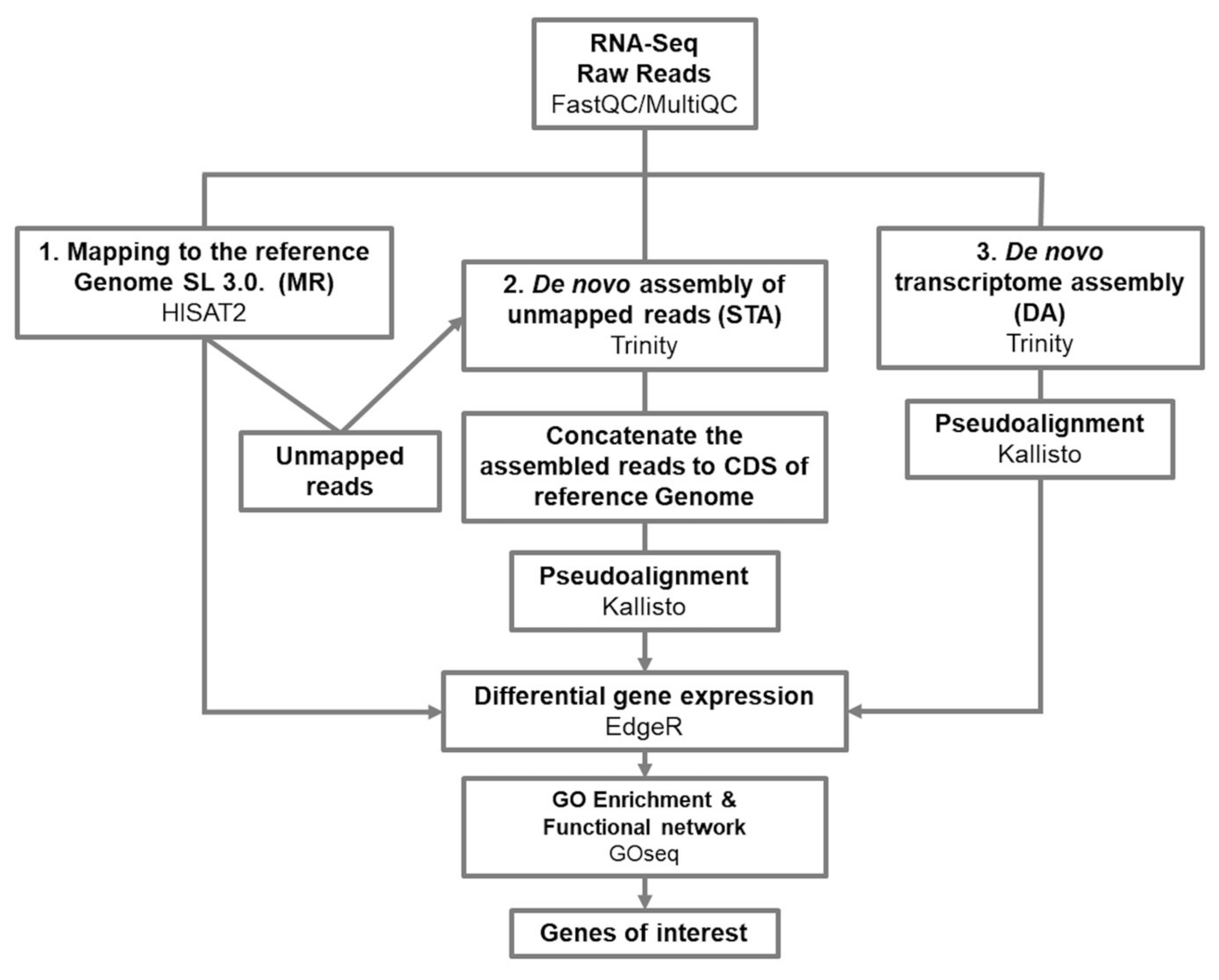
3.1. From Mapping to a Reference Genome, to a De Novo Assembly Transcriptome; a Complementary Approach
3.2. Global Transcriptional Profiling and Evidence of Resistance-Related Genes
4. Conclusions
5. Materials and Methods
5.1. Pathogenic Cmm Strain and Plant Material
5.2. Growth Conditions and Plant Infection
5.3. Cdna Library Preparation and RNA-Seq
5.4. Map to the S. lycopersicum Genome SL3.0
5.5. Map to the Semi De Novo Transcriptome
5.6. Map to the De Novo Transcriptome
5.7. Experimental Approach to Find Transcripts Showing Upregulation in S. arcanum Located at the Regions Spanning QTLs Associated with Resistance
5.8. Gene Expression Validation with qRT-PCR Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, E.F. A new tomato disease of economic importance. Science 1910, 31, 794–796. [Google Scholar]
- CABI. Clavibacter michiganensis subsp. michiganensis. In Invasive Species Compendium; CAB International: Wallingford, UK, 2021; Available online: www.cabi.org/isc (accessed on 1 September 2021).
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [Green Version]
- Borboa, F.J.; Rueda, P.E.O.; Acedo, F.E.; Ponce, J.F.; Cruz, M.; Juárez, O.G.; Ortega, A.M.G. Detección de Clavibacter michiganensis subespecies michiganensis en el tomate del estado de Sonora, México. Rev. Fitotec. Mex. 2009, 32, 319–326. [Google Scholar]
- Carlton, W.M.; Braun, E.J.; Gleason, M.L. Ingress of Clavibacter michiganensis subsp. michiganensis into tomato leaves through hydathodes. Phytopathology 1998, 88, 525–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontaxis, D.G. Leaf trichomes as avenues for infection by Corynebacterium michiganense. Phytopathology 1962, 52, 1306–1307. [Google Scholar]
- Strider, D.L. Bacterial canker of tomato caused by Corynebacterium michiganense. A literature review and bibliography. NC Agric. Exp. Stn. Tech. Bull. 1969, 193, 1–110. [Google Scholar]
- Gitaitis, R.D.; Beaver, R.W.; Voloudakis, A.E. Detection of Clavibacter michiganensis subsp. michiganensis in symptomless tomato transplants. Plant Dis. 1991, 75, 834–838. [Google Scholar] [CrossRef]
- Kleitman, F.; Barash, I.; Burger, A.; Iraki, N.; Falah, Y.; Sessa, G.; Weinthal, D.; Chalupowicz, L.; Gartemann, K.H.; Eichenlaub, R.; et al. Characterization of a Clavibacter michiganensis subsp. michiganensis population in Israel. Eur. J. Plant Pathol. 2008, 121, 463–475. [Google Scholar] [CrossRef]
- Gleason, M.L.; Gitaitis, R.D.; Ricker, M.D. Recent progress in understanding and controlling bacterial canker of tomato in eastern North America. Plant Dis. 1993, 77, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Strider, D.L. Survival studies with the tomato bacterial canker organism. Phytopathology 1967, 57, 1067–1071. [Google Scholar]
- Fatmi, M.; Schaad, N.W. Survival of Clavibacter michiganensis ssp. michiganensis in infected tomato stems under natural field conditions in California, Ohio and Morocco. Plant Pathol. 2002, 51, 149–154. [Google Scholar] [CrossRef]
- Hausbeck, M.; Bell, J.; Medina-Mora, C.; Podolsky, R.; Fulbright, D. Effect of bactericides on population sizes and spread of Clavibacter michiganensis subsp. michiganensis on tomatoes in the greenhouse and on disease development and crop yield in the field. Phytopathology 2000, 90, 38–44. [Google Scholar] [CrossRef] [Green Version]
- De León, L.; Siverio, F.; López, M.M.; Rodríguez, A. Comparative efficiency of chemical compounds for in vitro and in vivo activity against Clavibacter michiganensis subsp. michiganensis, the causal agent of tomato bacterial canker. Crop Prot. 2008, 27, 1277–1283. [Google Scholar] [CrossRef]
- Van Heusden, A.W.; Koornneef, M.; Voorrips, R.E.; Bruggemann, W.; Pet, G.; van Ginkel, R.V.; Chen, X.; Lindhout, P. Three QTLs from Lycopersicon peruvianum confer a high level of resistance to Clavibacter michiganensis ssp. michiganensis. Theor. Appl. Genet. 1999, 99, 1068–1074. [Google Scholar] [CrossRef]
- Francis, D.M.; Kabelka, E.; Bell, J.; Franchino, B.; St Clair, D. Resistance to bacterial canker in tomato (Lycopersicon hirsutum LA407) and its progeny derived from crosses to L. esculentum. Plant Dis. 2001, 85, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Kabelka, E.; Franchino, B.; Francis, D.M. Two loci from Lycopersicon hirsutum LA407 confer resistance to strains of Clavibacter michiganensis subsp. michiganensis. Phytopathology 2002, 92, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Sen, Y.; Zhu, F.; Vandenbroucke, H.; van der Wolf, J.; Visser, R.; van Heusden, A.W. Screening for new sources of resistance to Clavibacter michiganensis subsp. michiganensis (Cmm) in tomato. Euphytica 2013, 190, 309–317. [Google Scholar] [CrossRef]
- Balaji, V.; Mayrose, M.; Sherf, O.; Jacob-Hirsch, J.; Eichenlaub, R.; Iraki, N.; Manulis-Sasson, S.; Rechavi, G.; Barash, I.; Sessa, G. Tomato transcriptional changes in response to Clavibacter michiganensis subsp. michiganensis reveal a role for ethylene in disease development. Plant Physiol. 2008, 146, 1797–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Ávila, J.P.; Isordia-Jasso, M.I.; Castillo-Collazo, R.; Simpson, J.; Alpuche-Solís, Á.G. Gene expression analysis during interaction of tomato and related wild species with Clavibacter michiganensis subsp. michiganensis. Plant Mol. Biol. Rep. 2012, 30, 498–511. [Google Scholar] [CrossRef]
- Ameur, A.; Wetterbom, A.; Feuk, L.; Gyllensten, U. Global and unbiased detection of splice junctions from RNA-seq data. Genome Biol. 2010, 11, R34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozsolak, F.; Milos, P. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, F.; Wu, F.; Ané, C.; Tanksley, S.; Spooner, D.M. Do potatoes and tomatoes have a single evolutionary history, and what proportion of the genome supports this history? BMC Evol. Biol. 2009, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangiovanni, M.; Granata, I.; Thind, A.S.; Guarracino, M.R. From trash to treasure: Detecting unexpected contamination in unmapped NGS data. BMC Bioinform. 2019, 20, 168. [Google Scholar] [CrossRef]
- Honaas, L.A.; Wafula, E.K.; Wickett, N.J.; Der, J.P.; Zhang, Y.; Edger, P.P.; Altman, N.S.; Pires, J.C.; Leebens-Mack, J.H.; dePamphilis, C.W. Selecting superior de novo transcriptome assemblies: Lessons learned by leveraging the best plant genome. PLoS ONE 2016, 11, e0146062. [Google Scholar] [CrossRef] [PubMed]
- Hölzer, M.; Marz, M. De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-Seq assemblers. GigaScience 2019, 8, giz039. [Google Scholar] [CrossRef] [Green Version]
- Savidor, A.; Teper, D.; Gartemann, K.H.; Eichenlaub, R.; Chalupowicz, L.; Manulis-Sasson, S.; Barash, I.; Tews, H.; Mayer, K.; Giannone, R.J.; et al. The Clavibacter michiganensis subsp. michiganensis—tomato interactome reveals the perception of pathogen by the host and suggests mechanisms of infection. J. Proteome Res. 2012, 11, 736–750. [Google Scholar] [CrossRef]
- Basim, H.; Basim, E.; Tombuloglu, H.; Unver, T. Comparative transcriptome analysis of resistant and cultivated tomato lines in response to Clavibacter michiganensis subsp. michiganensis. Genomics 2021, 113, 2455–2467. [Google Scholar] [CrossRef] [PubMed]
- Balaji, V.; Smart, C.D. Over-expression of snakin-2 and extensin-like protein genes restricts pathogen invasiveness and enhances tolerance to Clavibacter michiganensis subsp. michiganensis in transgenic tomato (Solanum lycopersicum). Transgenic Res. 2012, 21, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Weber, H.; Vick, B.A.; Farmer, E.E. Dinor-oxo-phytodienoic acid: A new hexadecanoid signal in the jasmonate family. Proc. Natl. Acad. Sci. USA 1997, 94, 10473–10478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, H.; Chételat, A.; Caldelari, D.; Farmer, E.E. Divinyl ether fatty acid synthesis in late blight-diseased potato leaves. Plant Cell 1999, 11, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, S.; Weber, H.; Stolz, S.; Chételat, A.; Farmer, E.E. Fatty acid ketodienes and fatty acid ketotrienes: Michael addition acceptors that accumulate in wounded and diseased Arabidopsis leaves. Plant J. 2000, 24, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.E.; Ha, Y.I.; Choi, H.; Moon, J.Y.; Lee, J.; Shin, A.Y.; Park, C.J.; Yoon, G.M.; Kwon, S.Y.; Jo, I.H.; et al. Silencing of an α-dioxygenase gene, Ca-DOX, retards growth and suppresses basal disease resistance responses in Capsicum annum. Plant Mol. Biol. 2017, 93, 497–509. [Google Scholar] [CrossRef]
- Pertry, I.; Václavíková, K.; Depuydt, S.; Galuszka, P.; Spíchal, L.; Temmerman, W.; Stes, E.; Schmülling, T.; Kakimoto, T.; van Montagu, M.; et al. Identification of Rhodococcus fascians cytokinins and their modus operandi to reshape the plant. Proc. Natl. Acad. Sci. USA 2009, 106, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Huh, S.U.; Kojima, M.; Sakakibara, H.; Paek, K.; Hwang, I. The cytokinin-activated transcription factor ARR2 promotes plant immunity via TGA3/NPR1-dependent salicylic acid signaling in Arabidopsis. Dev. Cell 2010, 19, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Gómez, L.; Boller, T. FLS2: An LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 2000, 5, 1003–1011. [Google Scholar] [CrossRef]
- Choi, H.W.; Kim, Y.J.; Lee, S.C.; Hong, J.K.; Hwang, B.K. Hydrogen peroxide generation by the pepper extracellular peroxidase CaPO2 activates local and systemic cell death and defense response to bacterial pathogens. Plant Physiol. 2007, 145, 890–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Choi, H.W.; Hwang, B.K. The pepper E3 ubiquitin ligase RING1 gene, CaRING1, is required for cell death and the salicylic acid-dependent defense response. Plant Physiol. 2011, 156, 2011–2025. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, Y.; Ding, P.; Li, Y.; Kong, Q.; Zhang, Y. Splicing of receptor-like kinase-encoding SNC4 and CERK1 is regulated by two conserved splicing factors that are required for plant immunity. Mol. Plant 2014, 7, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Steffens, J.C. Overexpression of polyphenol oxidase in transgenic tomato plants results in enhanced bacterial disease resistance. Planta 2002, 215, 239–247. [Google Scholar] [CrossRef]
- Thipyapong, P.; Hunt, M.D.; Steffens, J.C. Antisense downregulation of polyphenol oxidase results in enhanced disease susceptibility. Planta 2004, 220, 105–117. [Google Scholar] [CrossRef]
- Li, G.C.; Jin, L.P.; Xie, K.Y.; Li, Y.; Qu, D.Y. Cloning of proteinase inhibitor gene StPI in diploid potato and its expression analysis. Agr. Sci. China 2007, 6, 1315–1321. [Google Scholar] [CrossRef]
- Charity, J.A.; Hughes, P.; Anderson, M.A.; Bittisnich, D.J.; Whitecross, M.; Higgins, T.J.V. Pest and disease protection conferred by expression of barley β-hordothionin and Nicotiana alata proteinase inhibitor genes in transgenic tobacco. Funct. Plant Biol. 2005, 32, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Mendy, B.; Wang’ombe, M.W.; Radakovic, Z.S.; Holbein, J.; Ilyas, M.; Chopra, D.; Holton, N.; Zipfel, C.; Grundler, F.M.; Siddique, S. Arabidopsis leucine-rich repeat receptor-like kinase NILR1 is required for induction of innate immunity to parasitic nematodes. PLoS Pathog. 2017, 13, e1006284. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, Y.; Yang, Z.; Xu, C.; Li, X.; Wang, S.; Zhang, Q. Xa26, a gene conferring resistance to Xanthomonas oryzae pv. oryzae in rice, encodes an LRR receptor kinase-like protein. Plant J. 2004, 37, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Godiard, L.; Sauviac, L.; Torii, K.U.; Grenon, O.; Mangin, B.; Grimsley, N.H.; Marco, Y. ERECTA, an LRR receptor-like kinase protein controlling development pleiotropically affects resistance to bacterial wilt. Plant J. 2003, 36, 353–365. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Ding, P.; Johnson, K.; Li, X.; Zhang, Y. The ankyrin-repeat transmembrane protein BDA1 functions downstream of the receptor-like protein SNC2 to regulate plant immunity. Plant Physiol. 2012, 159, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, X.; Ding, X.; Wang, Y.; Chen, Q.; Song, W.Y. The XA21 binding protein XB25 is required for maintaining XA21-mediated disease resistance. Plant J. 2013, 73, 814–823. [Google Scholar] [CrossRef]
- Li, G.; Chen, T.; Zhang, Z.; Li, B.; Tian, S. Roles of aquaporins in plant-pathogen interaction. Plants 2020, 9, 1134. [Google Scholar] [CrossRef]
- Tian, S.; Wang, X.; Li, P.; Wang, H.; Ji, H.; Xie, J.; Qiu, Q.; Shen, D.; Dong, H. Plant aquaporin AtPIP1; 4 links apoplastic H2O2 induction to disease immunity pathways. Plant Physiol. 2016, 171, 1635–1650. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Xie, Y.; Liu, G.; Lin, D.; He, C.; Shi, H. Molecular identification of GAPDHs in cassava highlights the antagonism of MeGAPCs and MeATG8s in plant disease resistance against cassava bacterial blight. Plant Mol. Biol. 2018, 97, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Laxalt, A.M.; Cassia, R.O.; Sanllorenti, P.M.; Madrid, E.A.; Andreu, A.B.; Daleo, G.R.; Conde, R.D.; Lamattina, L. Accumulation of cytosolic glyceraldehyde-3-phosphate dehydrogenase RNA under biological stress conditions and elicitor treatments in potato. Plant Mol. Biol. 1996, 30, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Huh, J.H.; Yu, Y.C.; Ho, L.H.; Chen, L.Q.; Tholl, D.; Frommer, W.B.; Guo, W.J. The Arabidopsis vacuolar sugar transporter SWEET2 limits carbon sequestration from roots and restricts Pythium infection. Plant J. 2015, 83, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- De Leone, M.J.; Hernando, C.E.; Romanowski, A.; Careno, D.A.; Soverna, A.F.; Sun, H.; Bologna, N.G.; Vázquez, M.; Schneeberger, K.; Yanovsky, M.J. Bacterial infection disrupts clock gene expression to attenuate immune responses. Curr. Biol. 2020, 30, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kong, J.; Cui, D.; Zhao, H.; Niu, Y.; Xu, M.; Jiang, G.; Zhao, Y.; Wang, W. Resistance against Ralstonia solanacearum in tomato depends on the methionine cycle and the γ-aminobutyric acid metabolic pathway. Plant J. 2019, 97, 1032–1047. [Google Scholar] [CrossRef]
- Shelp, B.J.; Zarei, A. Subcellular compartmentation of 4-aminobutyrate (GABA) metabolism in arabidopsis: An update. Plant Signal. Behav. 2017, 12, e1322244. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.F.; Zhang, S.C.; Yang, X.M.; Wang, C.P.; Huang, Q.Z.; Huang, R.-Z. Generation of a High-Density Genetic Map of Pepper (Capsicum annuum L.) by SLAF-seq and QTL Analysis of Phytophthora capsici Resistance. Horticulturae 2021, 7, 92. [Google Scholar] [CrossRef]
- Birch, P.R.; Avrova, A.O.; Duncan, J.M.; Lyon, G.D.; Toth, R.L. Isolation of potato genes that are induced during an early stage of the hypersensitive response to Phytophthora infestans. Mol. Plant Microbe Interact. 1999, 12, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Escobar-Sepúlveda, H.F.; Trejo-Téllez, L.I.; Pérez-Rodríguez, P.; Hidalgo-Contreras, J.V.; Gómez-Merino, F.C. Diacylglycerol kinases are widespread in higher plants and display inducible gene expression in response to beneficial elements, metal, and metalloid ions. Front. Plant Sci. 2017, 8, 129. [Google Scholar] [CrossRef] [Green Version]
- Arisz, S.A.; Testerink, C.; Munnik, T. Plant PA signaling via diacylglycerol kinase. Biochim. Biophys. Acta 2009, 1791, 869–875. [Google Scholar] [CrossRef]
- Kue Foka, I.C.; Ketehouli, T.; Zhou, Y.; Li, X.W.; Wang, F.W.; Li, H. The emerging roles of diacylglycerol kinase (DGK) in plant stress tolerance, growth, and development. Agronomy 2020, 10, 1375. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, J.; Zhang, H.; Song, F. Overexpression of a rice diacylglycerol kinase gene OsBIDK1 enhances disease resistance in transgenic tobacco. Mol. Cells 2008, 26, 258–264. [Google Scholar]
- Bittner-Eddy, P.D.; Crute, I.R.; Holub, E.B.; Beynon, J.L. RPP13 is a simple locus in Arabidopsis thaliana for alleles that specify downy mildew resistance to different avirulence determinants in Peronospora parasitica. Plant J. 2000, 21, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Roach, T.G.; Lång, H.K.; Xiong, W.; Ryhänen, S.J.; Capelluto, D.G. Protein trafficking or cell signaling: A dilemma for the adaptor protein TOM1. Front. Cell Dev. Biol. 2021, 9, 408. [Google Scholar] [CrossRef]
- Conlan, B.; Stoll, T.; Gorman, J.J.; Saur, I.; Rathjen, J.P. Development of a rapid in planta BioID system as a probe for plasma membrane-associated immunity proteins. Front. Plant Sci. 2018, 9, 1882. [Google Scholar] [CrossRef] [Green Version]
- Coaker, G.L.; Willard, B.; Kinter, M.; Stockinger, E.J.; Francis, D.M. Proteomic analysis of resistance mediated by Rcm 2.0 and Rcm 5.1, two loci controlling resistance to bacterial canker of tomato. Mol. Plant Microbe Interact. 2004, 17, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Le, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Sen, Y. Bacterial Canker Resistance in Tomato. Ph.D. Thesis, Wageningen University, Wageningen, The Netherlands, 2014. [Google Scholar]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Species. | 0–8 hpi | 8–24 hpi | Genes/Transcripts | ||
|---|---|---|---|---|---|
| Reference Genome SL3.0 | Up | Down | Up | Down | Total |
| S. lycopersicum | 1421 | 1483 | 295 | 245 | 14,477 FDR 0.01 |
| S. arcanum LA2157 | 1100 | 997 | 884 | 679 | |
| Semi de novo assembly | |||||
| S. lycopersicum | 1602 | 2249 | 500 | 277 | 24,741 FDR: 0.1 |
| S. arcanum LA2157 | 1687 | 1481 | 1315 | 1356 | |
| De novo assembly | |||||
| S. lycopersicum | 1409 | 1968 | 419 | 247 | 23,112 FDR: 0.1 |
| S. arcanum LA2157 | 1229 | 1244 | 1152 | 908 | |
| Average percentage of DEGs | |||||
| S. lycopersicum | 7.46 | 9.28 | 1.95 | 1.29 | |
| S. arcanum LA2157 | 6.57 | 6.08 | 5.46 | 4.69 | |
| p-value | 1.0 | 0.1 | 0.1 | 0.1 | |
| Gene/Transcript ID | Description | S. arc LogFC (0–8 hpi) | S. lyc LogFC (0–8 hpi) | S. arc LogFC (8–24 hpi) | S. lyc LogFC (8–24 hpi) |
|---|---|---|---|---|---|
| Reference genome | |||||
| Solyc06g074030.1 | CCR4-associated factor 1 homolog 9 | 3.14 * | −3.85 * | −2.16 | −1.07 |
| Solyc06g071280.3 | Protein EDS1B | 2.32 * | −1.66 * | −1.73 * | −0.46 |
| Solyc02g081500.3 | Rust resistance kinase Lr10 | 2.62 * | −1.87 * | −1.80 * | −0.23 |
| Solyc02g092140.1 | Protein PHLOEM PROTEIN 2-LIKE A10 | 5.37 * | 1.49 * | −2.96 * | −1.47 |
| Solyc08g074620.3 | Polyphenol oxidase E | 5.65 * | −0.38 | 0.07 | 3.03 |
| Solyc05g051530.3 | ABC transporter G family member 11 | 5.48 * | −0.04 | −4.90 * | −2.55 |
| Solyc11g017270.2 | Leucine-rich repeat receptor-like serine/threonine-protein kinase | 1.60 * | −1.20 * | −0.53 | 0.71 |
| Solyc08g080830.3 | Probable LRR receptor-like serine/threonine-protein kinase | 1.05 * | 0.48 | −0.69 * | −0.23 |
| Solyc12g009140.2 | Proteasome subunit α type-6-B | −0.83 * | −0.31 | 0.84 * | 0.22 |
| Solyc01g109590.3 | Probable receptor-like serine/threonine protein kinase | 2.65 * | 1.58 * | −2.16 * | −1.09 |
| Semi de novo assembly | |||||
| TRINITY_DN15795_c0_g2_i2 | Serine-type endopeptidase inhibitor activity | 6.19 * | −1.30 * | 0.97 | 0.04 |
| Solyc08g062360.3 | Ankyrin repeat-containing protein | 2.14 * | −2.67 * | −2.70 * | −0.88 |
| Solyc06g066370.3 | WRKY transcription factor WRKY24 | 1.33 * | −1.38 * | −0.96 | 0.51 |
| Solyc12g049330.2 | E3 ubiquitin-protein ligase SPL2 | 6.65 * | −0.05 | −1.15 | −0.06 |
| Solyc05g012900.3 | Putative late blight resistance protein homolog R1B-23 | 5.36 * | −1.62 | −1.03 | 0.85 |
| Solyc07g005090.3 | Class V chitinase | 2.84 * | 1.65 * | 0.83 | 0.07 |
| Solyc06g083390.3 | RPM1-interacting protein 4 | 1.29 * | −1.55 * | −0.74 | −0.31 |
| Solyc10g008010.3 | Proteasome subunit α type-2 | −0.67 | 0.10 | 1.02 * | −0.09 |
| Solyc12g007110.2 | Proline-rich receptor-like protein kinase PERK1 | 1.36 * | 0.44 | −0.81 * | 0.49 |
| De novo assembly | |||||
| TRINITY_DN22346_c2_g3_i1 | L-type lectin-domain containing receptor kinase | 2.44 * | −2.08 * | 0.91 | 2.82 * |
| TRINITY_DN19315_c2_g1_i2 | E3 ubiquitin-protein ligase RMA1H1 | 2.94 * | 1.84 | −0.75 | 0.89 |
| TRINITY_DN17399_c2_g1_i1 | NDR1/HIN1-like protein 2 | 4.54 | −3.19 * | −0.57 | −1.05 |
| TRINITY_DN16733_c1_g1_i7 | Annexin D4 | 2.72 * | −5.55 * | −1.32 | 1.74 |
| TRINITY_DN15770_c0_g1_i7 | Mitogen-activated protein kinase 3 | 2.12 * | −1.74 * | −0.54 | −0.35 |
| TRINITY_DN16314_c0_g3_i2 | NAC domain-containing protein 2 | 2.30 * | −2.04 * | −0.56 | 1.16 |
| TRINITY_DN22670_c0_g2_i2 | Leucine-rich repeat receptor-like serine/threonine-protein kinase | 1.67 | −6.48 * | −0.51 | 5.47 |
| TRINITY_DN16553_c1_g3_i1 | Wall-associated receptor kinase-like 20 | 3.20 * | 0.86 | −1.85 * | −0.05 |
| TRINITY_DN20729_c2_g1_i1 | Protein EDS1 | 2.09 * | −1.58 * | −1.83 * | −0.44 |
| TRINITY_DN14123_c3_g1_i2 | Probable receptor-like protein kinase | 1.75 * | 1.26 * | −2.12 * | −0.72 |
| Gene/Transcript ID | Description | S. arc LogFC |
|---|---|---|
| 0–8 hpi | ||
| TRINITY_DN14010_c2_g1_i5 | Tetraspanin-3 | 4.48 |
| TRINITY_DN24943_c4_g1_i9 | Linoleate 13S-lipoxygenase 2-1 | 3.88 |
| TRINITY_DN18887_c0_g4_i2 | UDP-glycosyltransferase 85A8 | 3.80 |
| TRINITY_DN16833_c0_g1_i15 | Unknown | 3.44 |
| TRINITY_DN13677_c0_g1_i8 | Bidirectional sugar transporter SWEET2a | 3.16 |
| TRINITY_DN23205_c0_g6_i3 | Ethylene-responsive transcription factor 1 | 2.95 |
| TRINITY_DN23169_c1_g1_i1 | γ aminobutyrate transaminase 2 | 2.78 |
| TRINITY_DN19385_c0_g1_i7 | Heavy metal-associated isoprenylated plant protein 32 | 2.63 |
| TRINITY_DN18065_c0_g1_i1 | Allene oxide synthase 2 chloroplastic | 2.48 |
| TRINITY_DN20602_c0_g1_i4 | Ethylene-responsive transcription factor 5 | 2.45 |
| 8–24 hpi | ||
| TRINITY_DN21371_c0_g1_i15 | Stromal 70 kDa heat shock-related protein | 6.39 |
| TRINITY_DN16003_c0_g1_i7 | Glyceraldehyde-3-phosphate dehydrogenase | 3.67 |
| TRINITY_DN15481_c0_g1_i3 | Aquaporin PIP2-1 | 3.26 |
| TRINITY_DN17153_c0_g1_i7 | Heat shock cognate 70 kDa protein 2 | 3.24 |
| TRINITY_DN23165_c1_g1_i17 | RuBisCO large subunit-binding protein subunit α | 3.15 |
| TRINITY_DN24736_c3_g1_i8 | Protein LNK1 | 2.74 |
| TRINITY_DN17225_c0_g1_i11 | Protein WVD2-like 7 | 2.7 |
| TRINITY_DN23140_c2_g1_i7 | Ferric reduction oxidase 6 | 2.65 |
| TRINITY_DN25011_c3_g1_i11 | Heat shock 70 kDa protein 15 | 2.52 |
| TRINITY_DN25230_c3_g1_i22 | Serine/arginine-rich splicing factor SC35 | 2.45 |
| Chromosome of QTL Spanning Region | Annotation | Transcript ID | S._arc LogFC | S._lyc LogFC | ||
|---|---|---|---|---|---|---|
| 0–8 hpi | 8–24 hpi | 0–8 hpi | 8–24 hpi | |||
| Ch5 | UPF0481 protein At3g47200 | TRINITY_DN21279_c1_g1_i2 | 6.90 | −1.33 | 0.35 | −1.33 |
| Ch5 | Hyoscyamine 6-dioxygenase | TRINITY_DN19945_c3_g1_i7 | 4.37 | 0.64 | 0.65 | 0.64 |
| Ch7 | Diacylglycerol kinase 7 | TRINITY_DN20884_c0_g1_i10 | 5.82 | 0.26 | 0.45 | −0.34 |
| Ch7 | Putative disease resistance RPP13-like protein 1 | TRINITY_DN22407_c0_g2_i3 | 1.69 | −1.12 | 0.21 | −0.30 |
| Ch9 | TOM1-like protein 6 | TRINITY_DN22806_c1_g1_i2 | 10.52 | −0.07 | −0.74 | 0.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereyra-Bistraín, L.I.; Ovando-Vázquez, C.; Rougon-Cardoso, A.; Alpuche-Solís, Á.G. Comparative RNA-Seq Analysis Reveals Potentially Resistance-Related Genes in Response to Bacterial Canker of Tomato. Genes 2021, 12, 1745. https://doi.org/10.3390/genes12111745
Pereyra-Bistraín LI, Ovando-Vázquez C, Rougon-Cardoso A, Alpuche-Solís ÁG. Comparative RNA-Seq Analysis Reveals Potentially Resistance-Related Genes in Response to Bacterial Canker of Tomato. Genes. 2021; 12(11):1745. https://doi.org/10.3390/genes12111745
Chicago/Turabian StylePereyra-Bistraín, Leonardo I., Cesaré Ovando-Vázquez, Alejandra Rougon-Cardoso, and Ángel G. Alpuche-Solís. 2021. "Comparative RNA-Seq Analysis Reveals Potentially Resistance-Related Genes in Response to Bacterial Canker of Tomato" Genes 12, no. 11: 1745. https://doi.org/10.3390/genes12111745