
A Comprehensive Study of the Genus Sanguisorba (Rosaceae) Based on the Floral Micromorphology, Palynology, and Plastome Analysis




Abstract
:1. Introduction
2. Results
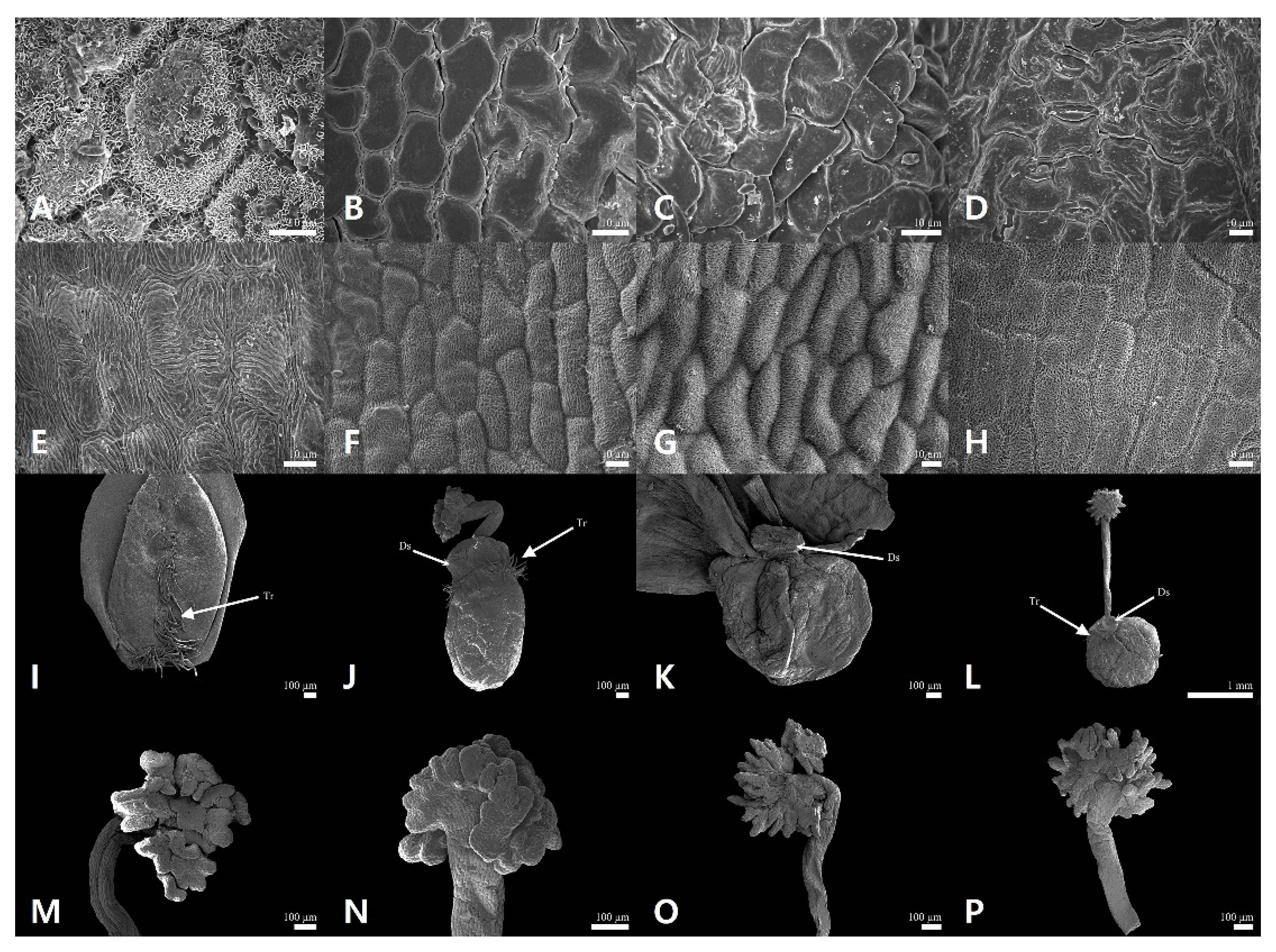
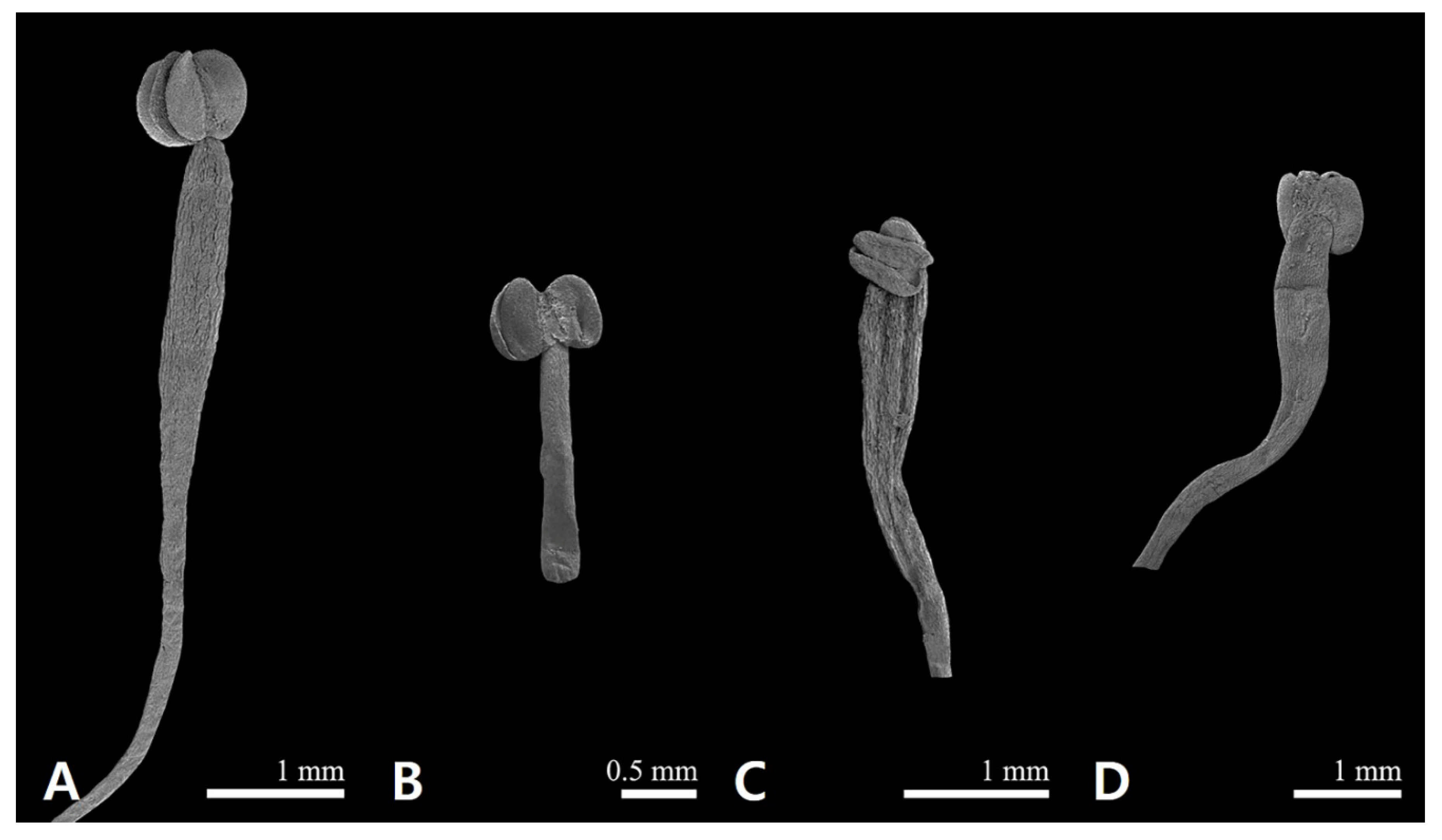
2.1. Floral Micromorphological Characteristics of Sanguisorba
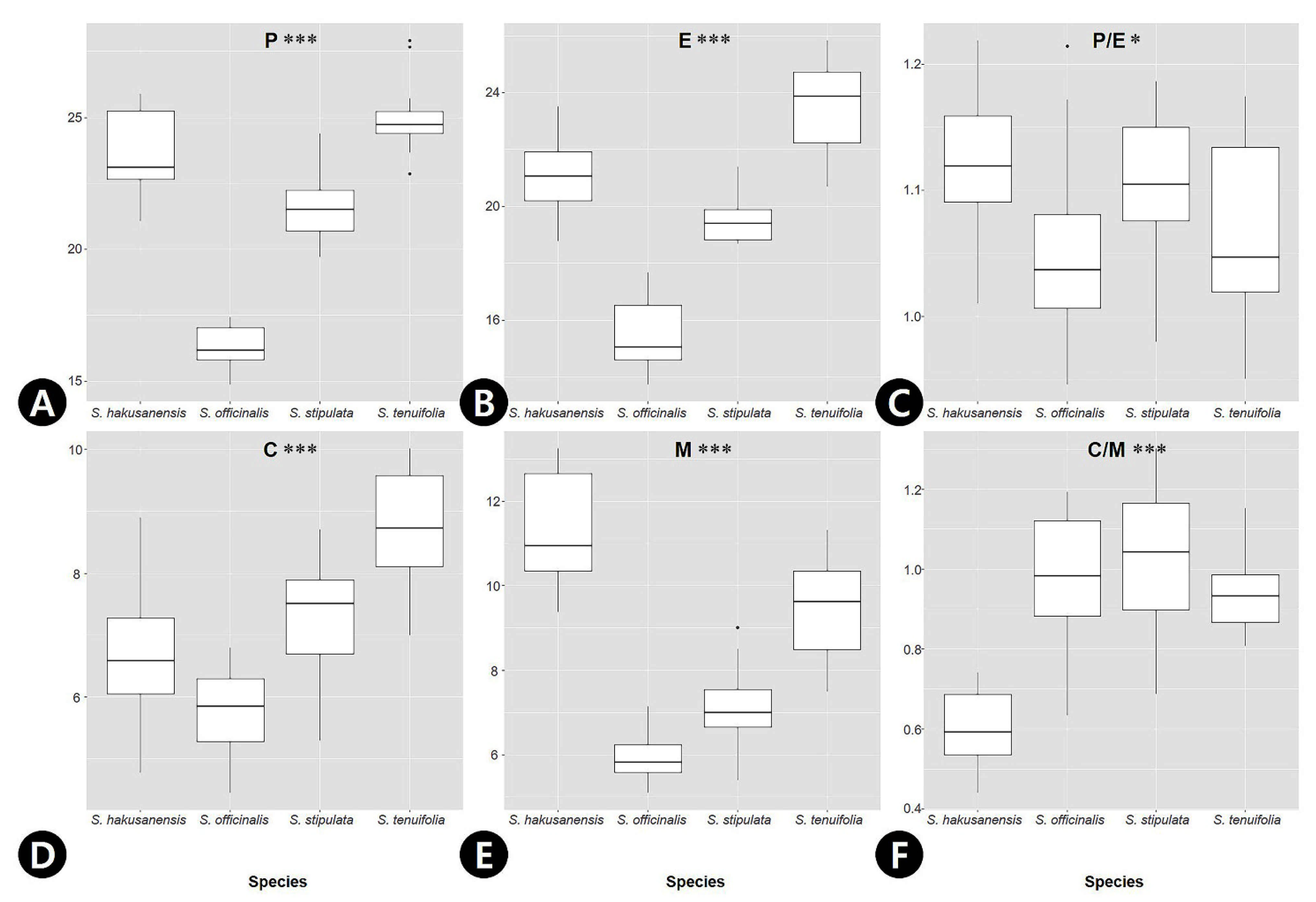
2.2. Palynological Characteristics of Sanguisorba
| 1. | Outer sepal waxes platelets; hypanthium trichomes basal and ventral part; tricolporate pollen grains; mesocolpus wider than colpus | S. hakusanensis |
| 1′. | Outer sepal waxes absent; hypanthium trichomes upper part or absent; hexacolporate pollen grains; mesocolpus and colpus almost equal | 2 |
| 2. | Filament length 1–3 mm and filiform shaped; hypanthium ellipsoid; stigma fimbriae blunted apex | S. officinalis |
| 2′. | Filament length 5–8 mm and compressed-dilated; hypanthium globose; stigma fimbriae acute apex | 3 |
| 3. | Hypanthium trichomes absent | S. stipulata |
| 3′. | Hypanthium trichomes upper part | S. tenuifolia |
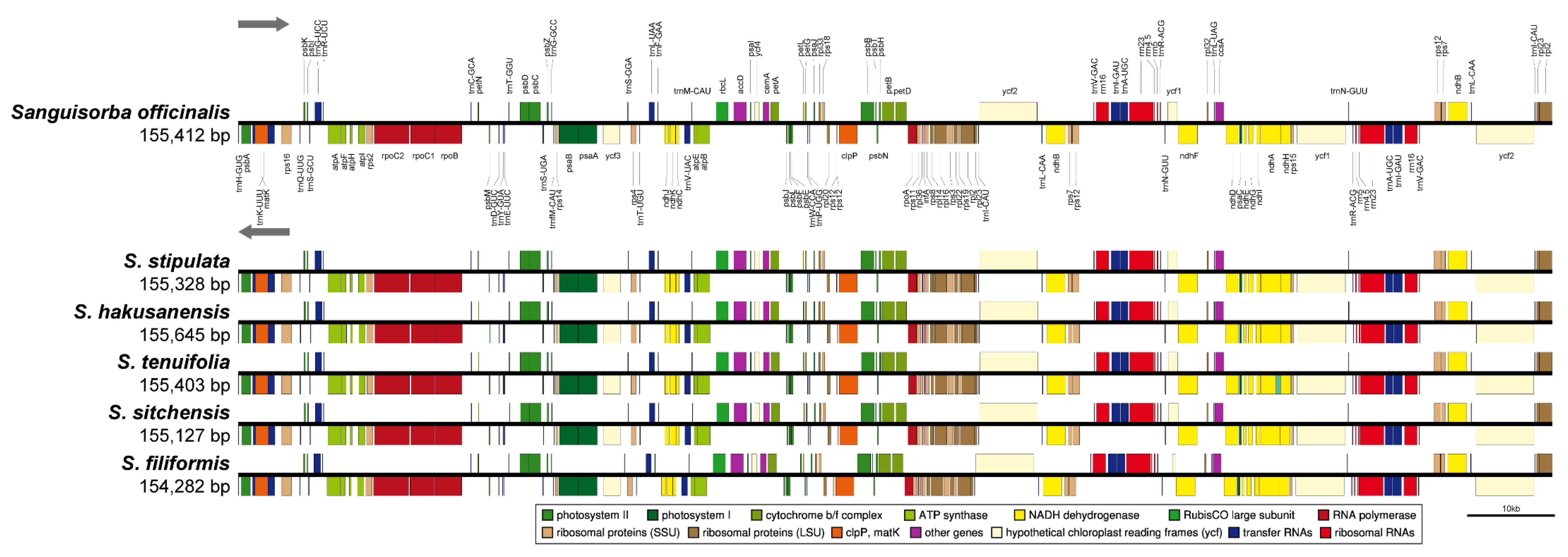
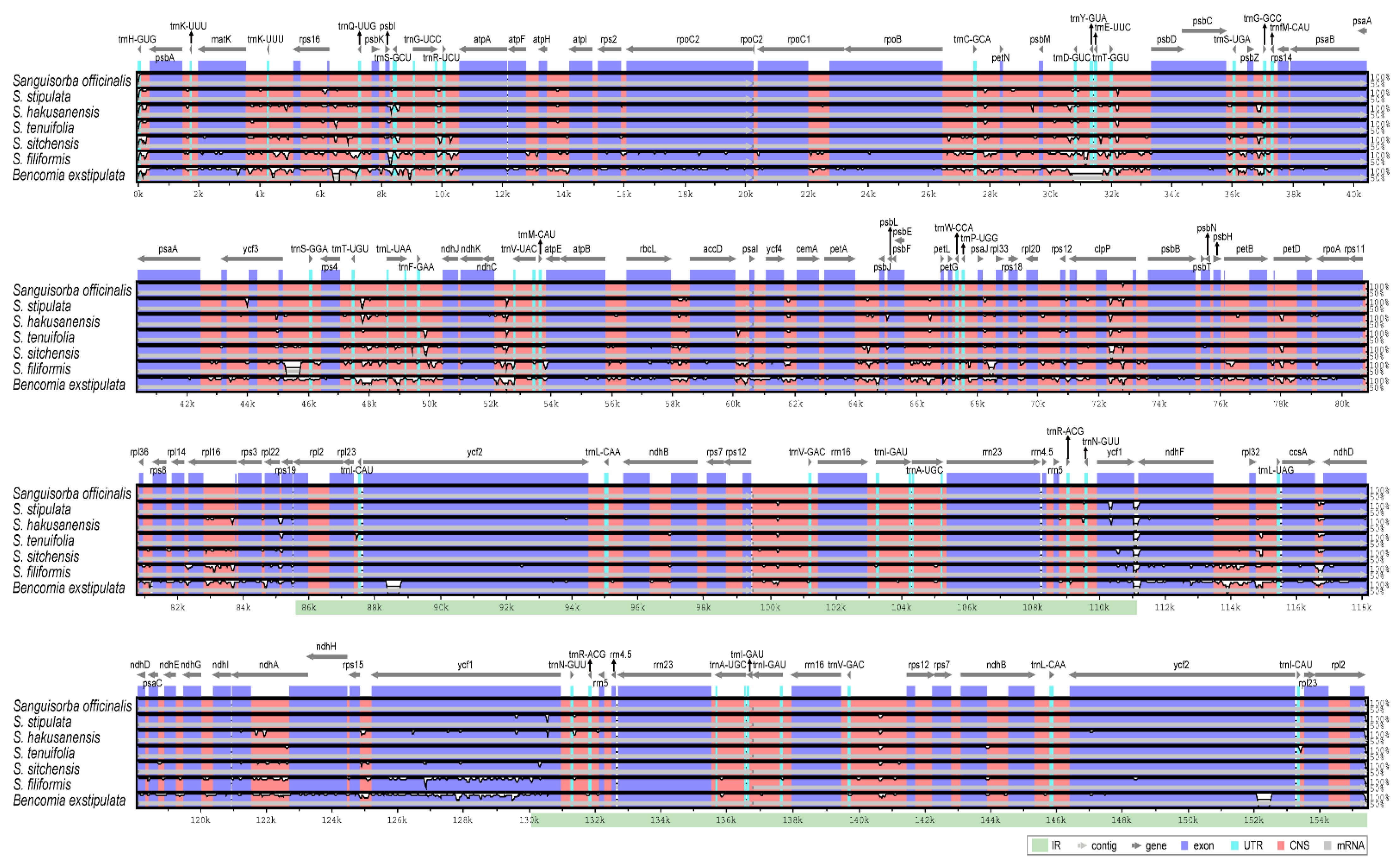
2.3. The Chloroplast Genome Organization of Sanguisorba
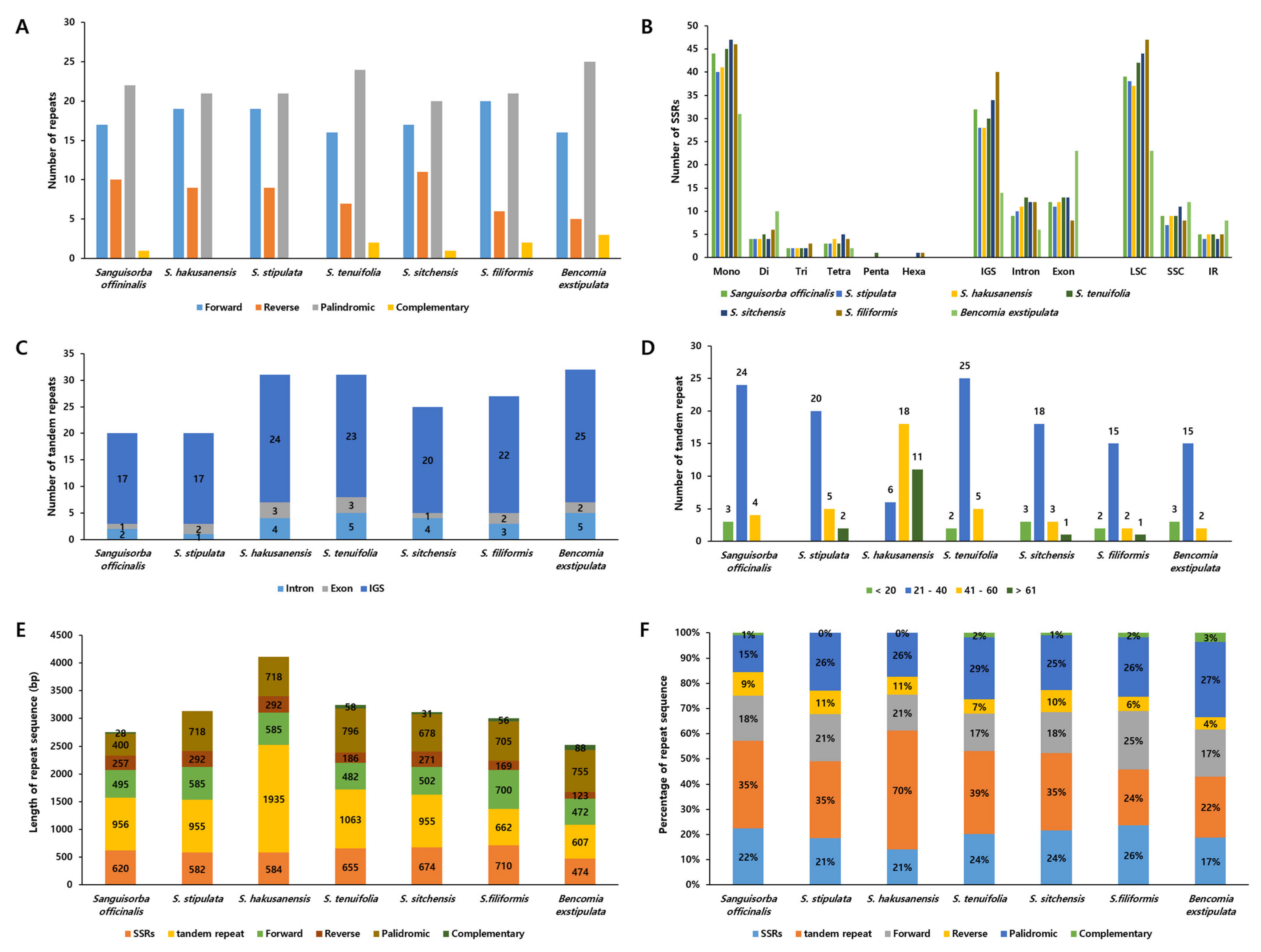
2.4. The Repeat Sequences of the Sanguisorba Chloroplast Genomes
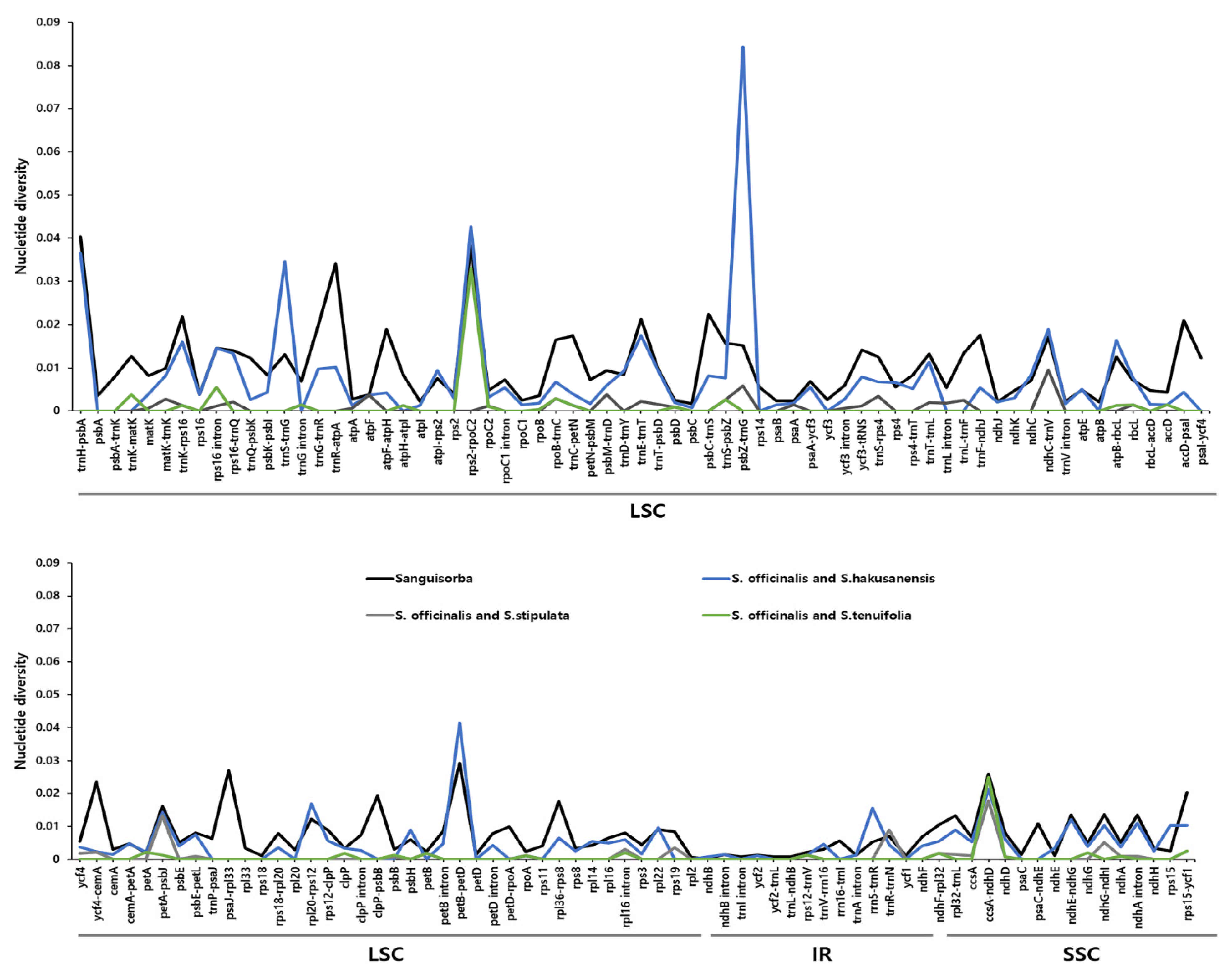
2.5. Comprehensive Comparative Analysis of the Sanguisorba Chloroplast Genomes
2.6. New DNA Barcode for the Authentication of Sanguisorbae Radix
2.7. The Phylogenetic Relationships among Sanguisorba
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Microscopic Observations
4.3. Genome Sequencing and Assembly
4.4. Genome Annotation and Comparative Analysis
4.5. Repeat Analysis
4.6. Development of New DNA Barcodes for Distinguishing Sanguisorba
4.7. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schulze-Menz, G.K. Rosaceae. In Engler’s Syllabus der Pflanzenfamilien II, 12th ed.; Melchior, H., Ed.; Gebrüder Borntraeger: Berlin, Germany, 1964; pp. 209–218. [Google Scholar]
- Nordborg, G. Sanguisorba L. Sarcopoterium Spach, and Bencomia Webb et Berth. delimitation and subdivision of the genera. Opera Bot. 1966, 11, 1–103. [Google Scholar]
- Mabberley, D.J. The Plant-Book: A Portable Dictionary of the Vascular Plants; Cambridge University Press: New York, NY, USA, 1997. [Google Scholar]
- Li, C.L.; Ikeda, H.; Ohba, H. Sanguisorba L. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2003; pp. 384–387. [Google Scholar]
- The International Plant Names Index and World Checklist of Selected Plant Families. 2021. Available online: http://www.ipni.org and http://apps.kew.org/wcsp/ (accessed on 4 July 2021).
- Lee, J.; Lee, B.Y.; Kim, Y.S. A taxonomic study of Korean taxa of the Rosaceae genus Sanguisorba. Korean J. Plant Taxon. 2000, 30, 269–285. [Google Scholar] [CrossRef]
- Naruhashi, N. Sanguisorba L. In Flora of Japan IIb. Angiospermae Dicotyledoneae Archichlamydeae (b); Iwatsuki, K., Boufford, D.E., Ohba, H., Eds.; Kodansha: Tokyo, Japan, 2001; pp. 180–185. [Google Scholar]
- Lee, B.Y. Sanguisorba L. In The Genera of Vascular Plants of Korea; Flora of Korean Editorial Committee, Ed.; Hongreung Publishing Co.: Seoul, Korea, 2018; pp. 731–733. [Google Scholar]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; Medical Science and Technology Press: Beijing, China, 2015. [Google Scholar]
- Korea Food and Drug Administration. The Korean Herbal Pharmacopoeia, 4th ed.; The KFDA Notification: Seoul, Korea, 2020.
- Korea Institute of Oriental Medicine [KIOM]. Defining Dictionary for Medicinal Herbs. Available online: http://boncho.kiom.re.kr/codex (accessed on 4 July 2021).
- Tu, J.; Li, Q.; Zhou, B. The Tannins from Sanguisorba officinalis L. (Rosaceae): A systematic study on the metabolites of rats based on HPLC–LTQ–Orbitrap MS2 analysis. Molecules 2021, 26, 4053. [Google Scholar] [CrossRef] [PubMed]
- Gawron-Gzella, A.; Witkowska-Banaszczak, E.; Bylka, W.; Dudek-Makuch, M.; Odwrot, A.; Skrodzka, N. Chemical composition, antioxidant and antimicrobial activities of Sanguisorba officinalis L. extracts. Pharm. Chem. J. 2016, 50, 244–249. [Google Scholar] [CrossRef]
- Chen, X.; Li, B.G.; Gao, Y.; Ji, J.X.; Wu, Z.L.; Chen, S. Saponins from Sanguisorba officinalis improve hematopoiesis by promoting survival through FAK and Erk1/2 activation and modulating cytokine production in bone marrow. Front. Pharmacol. 2017, 8, 130. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.; Inn, K.S.; Jang, Y.P.; Lee, K.T.; Lee, J.H. Phytotherapeutic activities of Sanguisorba officinalis and its chemical constituents: A review. Am. J. Chin. Med. 2018, 46, 299–318. [Google Scholar] [CrossRef]
- Su, X.D.; Guo, R.H.; Yang, S.Y.; Kim, Y.H.; Kim, Y.R. Anti-bacterial effects of components from Sanguisorba officinalis L. on Vibrio vulnificus and their soluble epoxide hydrolase inhibitory activity. Nat. Prod. Res. 2019, 33, 3445–3449. [Google Scholar] [CrossRef]
- Chung, K.-S.; Elisens, W.J.; Skvarla, J.J. Pollen morphology and its phylogenetic significance in tribe Sanguisorbeae (Rosaceae). Plant Syst. Evol. 2010, 285, 139–148. [Google Scholar] [CrossRef]
- Lee, S.T.; Heo, K.I.; Cho, J.H.; Lee, C.H.; Chen, W.; Kim, S.C. New insights into pollen morphology and its implications in the phylogeny of Sanguisorba L. (Rosaceae; Sanguisorbeae). Plant Syst. Evol. 2011, 291, 227–242. [Google Scholar] [CrossRef]
- Wang, J.R.; Wang, X.; Su, N.; Li, Q.J.; Zhang, X.H.; Ma, Y.P.; Zhao, L.; Toni, J.F.G.; De Craene, L.R. Floral morphology and morphogenesis in Sanguisorba (Rosaceae): Flower diversification despite petal reduction and spatial constraints. Bot. J. Linn. Soc. 2020, 193, 47–63. [Google Scholar] [CrossRef]
- Çildir, H.; Kahraman, A.; Dogan, M. Petal and sepal epidermal micromorphology of six Lathyrus taxa (Fabaceae) and their systematic value. Not. Bot. Horti Agrobot. Cluj-Napoca 2012, 40, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Song, J.H.; Hong, S.P. A study on the petal micromorphological characteristics of the tribe Sorbarieae (Rosaceae). Korean J. Plant Res. 2016, 29, 376–384. [Google Scholar] [CrossRef]
- Song, J.H.; Roh, H.S.; Hong, S.P. Petal micromorphology and its systematic implications in Rosaceae tribe Spiraeeae. Brittonia 2020, 72, 111–122. [Google Scholar] [CrossRef]
- Park, I.; Yang, S.; Song, J.H.; Moon, B.C. Dissection for floral micromorphology and plastid genome of valuable medicinal borages Arnebia and Lithospermum (Boraginaceae). Front. Plant Sci. 2020, 11, 606463. [Google Scholar] [CrossRef]
- Wrońska-Pilarek, D.; Jagodziński, A.M. Systematic importance of pollen morphological features of selected species from the genus Rosa (Rosaceae). Plant Syst. Evol. 2011, 295, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.H.; Zhou, X.M.; Li, M.; Xu, B.; Deng, H.N.; Yu, Q.; Gao, X.F. Pollen morphology in Rubus (Rosaceae) and its taxonomic implications. Plant Syst. Evol. 2019, 305, 705–716. [Google Scholar] [CrossRef]
- Lechowicz, K.; Wrońska-Pilarek, D.; Bocianowski, J.; Maliński, T. Pollen morphology of Polish species from the genus Rubus L. (Rosaceae) and its systematic importance. PLoS ONE 2020, 15, e0221607. [Google Scholar] [CrossRef]
- Reitsma, T. Pollen morphology of some European Rosaceae. Acta Bot. Neerl. 1966, 15, 290–307. [Google Scholar] [CrossRef] [Green Version]
- Eide, F. Key for northwest European Rosaceae pollen. Grana 1981, 20, 101–118. [Google Scholar] [CrossRef]
- Hebda, R.J.; Chinnappa, C.C. Studies on pollen morphology of Rosaceae. Acta Bot. Gallica 1994, 141, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.H.; Wei, Z.X.; Wu, Z.Y. Pollen morphology of Maloideae of China (Rosaceae). Acta Bot. Yunnan. 2000, 22, 47–52. [Google Scholar]
- Song, J.H.; Moon, H.K.; Hong, S.P. Pollen morphology of the tribe Sorbarieae (Rosaceae). Plant Syst. Evol. 2016, 302, 853–869. [Google Scholar] [CrossRef]
- Song, J.H.; Moon, H.K.; Oak, M.K.; Hong, S.P. Phylogenetic evaluation of pollen and orbicule morphology in Rosaceae tribe Neillieae (subfamily Amygdaloideae). Bot. J. Linn. Soc. 2017, 183, 439–453. [Google Scholar] [CrossRef]
- Song, J.H.; Oak, M.K.; Roh, H.S.; Hong, S.P. Morphology of pollen and orbicules in the tribe Spiraeeae (Rosaceae) and its systematic implications. Grana 2017, 56, 351–367. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, R.; Knoop, V. Genomics of Chloroplasts and Mitochondria; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar]
- Song, Y.; Dong, W.P.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef] [Green Version]
- Park, I.; Yang, S.; Kim, W.J.; Noh, P.; Lee, H.O.; Moon, B.C. The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic Pharbitidis Semen (Seeds of I. nil or I. purpurea). Front. Plant Sci. 2018, 9, 965. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.C.; Lee, J.; Lee, H.O.; Joh, H.J.; Kim, N.H.; Park, H.S.; Yang, T.J. Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLoS ONE 2015, 10, e0117159. [Google Scholar] [CrossRef]
- Park, I.; Kim, W.J.; Yang, S.; Yeo, S.M.; Li, H.; Moon, B.C. The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS ONE 2017, 12, e0184257. [Google Scholar]
- Park, I.; Yang, S.; Kim, W.J.; Noh, P.; Lee, H.O.; Moon, B.C. Complete chloroplast genome of Sanguisorba x tenuifolia Fisch ex Link. Mitochondrial DNA Part B 2018, 3, 909–910. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.X.; Xian, Y.F.; Xiang, L.; Zhang, D.; Shi, Y.H.; Wu, M.L.; Dong, G.Q.; Ip, S.P.; Lin, Z.X.; Wu, L.; et al. Complete chloroplast genomes from Sanguisorba: Identity and variation among four species. Molecules 2018, 23, 2137. [Google Scholar] [CrossRef] [Green Version]
- Costa, V.B.S.; Pimentel, R.M.M.; Chagas, M.G.S.; Alves, G.D.; Castro, C.C. Petal micromorphology and its relationship to pollination. Plant Biol. 2017, 19, 115–122. [Google Scholar] [CrossRef]
- Barthlott, W. Epicuticular wax ultrastructure and systematics. In Caryophyllales: Evolution and Systematics; Behnke, H.D., Mabry, T.J., Eds.; Springer: Berlin, Germany, 1994; pp. 75–86. [Google Scholar]
- Barthlott, W.; Neinhuis, C.; Cutler, D.; Ditsch, F.; Meusel, I.; Theisen, I. Classification and terminology of plant epicuticular waxes. Bot. J. Linn. Soc. 1998, 126, 237–260. [Google Scholar] [CrossRef]
- Barthlott, W.; Theisen, I.; Borsch, T.; Neinhuis, C. Epicuticular waxes and vascular plant systematics: Integrating micromorphological and chemical data. In Deep Morphology: Toward a Renaissance of Morphology in Plant Systematics; Stuessy, T.F., Mayer, V., Hörandl, E., Eds.; ARG Gantner Verlag: Ruggell, Germany, 2003; pp. 189–206. [Google Scholar]
- Wissemann, V. Epicuticular wax morphology and the taxonomy of Rosa (section Caninae, subsection Rubiginosae). Plant Syst. Evol. 2000, 221, 107–112. [Google Scholar] [CrossRef]
- Jeffree, C.E. The fine structure of the plant cuticle. In Biology of the Plant Cuticle. Annual Plant Reviews; Riederer, M., Müller, C., Eds.; Blackwell: Des Moines, IA, USA, 2006; Volume 23, pp. 11–125. [Google Scholar]
- Tomaszewski, D.; Zieliński, J. Epicuticular wax structures on stems and comparison between stems and leaves—A survey. Flora-Morphol. Distrib. Funct. Ecol. Plants 2014, 209, 215–232. [Google Scholar] [CrossRef]
- Tomaszewski, D.; Byalt, A.; Gawlak, M. Leaf and stem epicuticular wax structures in Lonicera species (Caprifoliaceae). Nord. J. Bot. 2019, 37, e02210. [Google Scholar] [CrossRef]
- Juhari, A.A.A.; Talip, N.; Che, C.N.A.; Amri, S.K.A.R.; Mansur, N.S. Taxonomic significance of petal and sepal micromorphological characteristics in some Justicia nees (Acanthaceae) species in Peninsular Malaysia. Trop. Life Sci. Res. 2019, 30, 51–69. [Google Scholar] [CrossRef]
- Besi, E.E.; Eee, L.Y.; Eng, K.H.; Ching, T.M.; Nulit, R.; Go, R. Floral-surface micromorphology of Corybas selangorensis J. Dransf. & G. Sm. and Corybas holttumii J. Dransf. & G. Sm. (Orchidaceae). J. Orchid Soc. India 2019, 33, 47–56. [Google Scholar]
- Besi, E.E.; Nikong, D.; Mustafa, M.; Yong, C.S.Y.; Go, R. Taxonomic placement of four confusable Crepidium species (Orchidaceae, Malaxidinae) based on macro-and micro-morphological analyses, including a note on two new records to Peninsular Malaysia. Phytotaxa 2020, 454, 31–44. [Google Scholar] [CrossRef]
- Besi, E.E.; Jia, L.S.; Mustafa, M.; Yong, C.S.; Go, R. Comparative floral surface micromorphology helps to discriminate between species of Paphiopedilum (Orchidaceae: Cypripedioideae) from Peninsular Malaysia. Lankesteriana 2021, 21, 17–31. [Google Scholar]
- Koch, K.; Barthlott, W. Plant epicuticular waxes: Chemistry, form, self-assembly and function. Nat. Prod. Commun. 2006, 1, 1067–1072. [Google Scholar] [CrossRef]
- Schreiber, L. Characterisation of polar paths of transport in plant cuticles. In Biology of the Plant Cuticle. Annual Plant Reviews; Riederer, M., Müller, C., Eds.; Blackwell: Des Moines, IA, USA, 2006; Volume 23, pp. 280–289. [Google Scholar]
- Haworth, M.; McElwain, J. Hot, dry, wet, cold or toxic? Revisiting the ecological significance of leaf and cuticular micromorphology. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2008, 262, 79–90. [Google Scholar] [CrossRef]
- Whitehead, D.R. Wind pollination in the angiosperms: Evolutionary and environmental considerations. Evolution 1969, 23, 28–35. [Google Scholar] [CrossRef]
- Niklas, K.J. The aerodynamics of wind pollination. Bot. Rev. 1985, 51, 328–386. [Google Scholar] [CrossRef]
- Hebda, R.J.; Chinnappa, C.C. Studies on pollen morphology of Rosaceae in Canada. Rev. Palaeobot. Palynol. 1990, 64, 103–108. [Google Scholar] [CrossRef]
- Zhou, L.H.; Wei, Z.X.; Wu, Z.Y. Pollen morphology of Prunoideae of China (Rosaceae). Acta Bot. Yunnan. 1999, 21, 207–211. [Google Scholar]
- Wang, F.X.; Qian, N.F.; Zhang, Y.L.; Yang, H.Q. Pollen Flora of China; Science Press: Beijing, China, 1997. [Google Scholar]
- Lee, F.; Wan, P.; Gao, S.; Ding, Y.; Zhou, F. Pollen morphology identification of five species of medicinal Sanguisorba with electron microscope. China J. Chin. Mater. Med. 1999, 24, 715–717. [Google Scholar]
- Xu, Q.H. Pollen Morphology of Common Cultivated Plants in China—A Reference for Looking for Human Traces in the Stratum; Science Press: Beijing, China, 2005. [Google Scholar]
- Punt, W.; Hoen, P.P.; Blackmore, S.; Nilsson, S.; Le Thomas, A. Glossary of pollen and spore terminology. Rev. Palaeobot. Palynol. 2007, 143, 1–81. [Google Scholar] [CrossRef]
- Agashe, S.N.; Caulton, E. Pollen and Spores: Applications with Special Emphasis on Aerobiology and Allergy; Science Publishers: Grafton County, NH, USA, 2009. [Google Scholar]
- El-Ghazaly, G.; Nilsson, S. Development of tapetum and orbicules of Catharanthus roseus (Apocynaceae). In Pollen Spores: Patterns of Diversity; Balckmore, S., Barnes, S.H., Eds.; Clarendon Press: Oxford, UK, 1991; pp. 317–329. [Google Scholar]
- Huysmans, S.; El-Ghazaly, G.; Smets, E. Orbicules: Still a well hidden secret of the anther. In Plant Systematics for the 21st Century, Wenner-Gren International Series; Nordenstam, B., El-Ghazaly, G., Kassas, M., Eds.; Portland Press: London, UK, 2000; Volume 77, pp. 201–212. [Google Scholar]
- Galati, B.G. Ubisch bodies in angiosperms. In Advances in Plant Reproductive Biology; Chauhann, M.R., Pandey, A.K., Eds.; Narendra Publishing House: Delhi, India, 2003; pp. 1–20. [Google Scholar]
- Moon, H.K. The phylogenetic potential of orbicules in angiosperms. Korean J. Plant Taxon. 2018, 48, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Gichira, A.W.; Li, Z.; Saina, J.K.; Long, Z.; Hu, G.; Gituru, R.W.; Wang, Q.; Chen, J. The complete chloroplast genome sequence of an endemic monotypic genus Hagenia (Rosaceae): Structural comparative analysis, gene content and microsatellite detection. PeerJ 2017, 5, e2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, K.I.; Park, J.; Xi, H.; Min, J. The complete chloroplast genome of Agrimonia pilosa Ledeb. isolated in Korea (Rosaceae): Investigation of intraspecific variations on its chloroplast genomes. Mitochondrial DNA Part B 2020, 5, 2264–2266. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhan, D.F.; Jia, X.; Mei, W.L.; Dai, H.F.; Chen, X.T.; Peng, S.Q. Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the Malvales order. Front. Plant Sci. 2016, 7, 280. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.; Li, Y.; Wang, W. Selection shapes the patterns of codon usage in three closely related species of genus Misgurnus. Genomics 2018, 110, 134–142. [Google Scholar] [CrossRef]
- Ivanova, Z.; Sablok, G.; Daskalova, E.; Zahmanova, G.; Apostolova, E.; Yahubyan, G.; Baev, V. Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 2017, 8, 204. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.H.; Shang, A.Q.; Zhang, S.; Yu, X.Y.; Ren, Y.C.; Yang, M.S.; Wang, J.M. The first complete chloroplast genome sequences of Ulmus species by de novo sequencing: Genome comparative and taxonomic position analysis. PLoS ONE 2017, 12, e0171264. [Google Scholar] [CrossRef] [Green Version]
- Delannoy, E.; Fujii, S.; Colas des Francs-Small, C.; Brundrett, M.; Small, I. Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 2011, 28, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, K.H.; Morden, C.W.; Palmer, J.D. Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant. Proc. Natl. Acad. Sci. USA 1992, 89, 10648–10652. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.Y.; Yang, J.X.; Li, H.K.; Zhao, H.S. Chloroplast genomes of two species of Cypripedium: Expanded genome size and proliferation of AT-biased repeat sequences. Front. Plant Sci. 2021, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, R.; Van der Bank, M.; Bogarin, D.; Warner, J.; Pupulin, F.; Gigot, G.; Maurin, O.; Duthoit, S.; Barraclough, T.G.; Savolainen, V. DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. USA 2008, 105, 2923–2928. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of Orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.X.; Wang, Y.W.; He, P.Z.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C.X. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef]
- Smidt, E.D.; Paez, M.Z.; Vieira, L.N.; Viruel, J.; de Baura, V.A.; Balsanelli, E.; de Souza, E.M.; Chase, M.W. Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PLoS ONE 2020, 15, e0227991. [Google Scholar]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.P.; Chuang, W.J.; Huang, S.S.F.; Hwang, S.Y. Evidence for the existence of some dissociation in an otherwise strong linkage disequilibrium between mitochondrial and chloroplastic genomes in Cyclobalanopsis glauca. Mol. Ecol. 2003, 12, 2661–2668. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Deng, Y.; Wang, J. The complete chloroplast genomes of Echinacanthus species (Acanthaceae): Phylogenetic relationships, adaptive evolution, and screening of molecular markers. Front. Plant Sci. 2018, 9, 1989. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef] [Green Version]
- Li, P.P.; Lou, G.L.; Cai, X.R.; Zhang, B.; Cheng, Y.Q.; Wang, H.W. Comparison of the complete plastomes and the phylogenetic analysis of Paulownia species. Sci. Rep. 2020, 10, 2225. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.N.; Milne, R.I.; Du, X.Y.; Liu, J.; Wu, Z.Y. Characteristics and mutational hotspots of plastomes in Debregeasia (Urticaceae). Front. Genet. 2020, 11, 729. [Google Scholar] [CrossRef]
- Chen, S.L.; Yao, H.; Han, J.P.; Liu, C.; Song, J.Y.; Shi, L.C.; Zhu, Y.J.; Ma, X.Y.; Gao, T.; Pang, X.H.; et al. Validation of the ITS2 Region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 2010, 5, e8613. [Google Scholar] [CrossRef]
- Selvaraj, D.; Shanmughanandhan, D.; Sarma, R.K.; Joseph, J.C.; Srinivasan, R.V.; Ramalingam, S. DNA barcode ITS effectively distinguishes the medicinal plant Boerhavia diffusa from its adulterants. Genom. Proteom. Bioinform. 2012, 10, 364–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wu, X.; Liu, C.; Newmaster, S.; Ragupathy, S.; Kress, W.J. Progress in the use of DNA barcodes in the identification and classification of medicinal plants. Ecotox. Environ. Safe. 2021, 208, 111691. [Google Scholar] [CrossRef]
- Chung, H.Y.; Won, S.Y.; Kim, Y.K.; Kim, J.S. Development of the chloroplast genome-based InDel markers in Niitaka (Pyrus pyrifolia) and its application. Plant Biotechnol. Rep. 2019, 13, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Hechanova, S.L.; Bhattarai, K.; Simon, E.V.; Clave, G.; Karunarathne, P.; Ahn, E.K.; Li, C.P.; Lee, J.S.; Kohli, A.; Hamilton, N.R.S.; et al. Development of a genome-wide InDel marker set for allele discrimination between rice (Oryza sativa) and the other seven AA-genome Oryza species. Sci. Rep. 2021, 11, 8962. [Google Scholar] [CrossRef]
- Mishima, M.; Ohmido, N.; Fukui, K.; Yahara, T. Trends in site-number change of rDNA loci during polyploid evolution in Sanguisorba (Rosaceae). Chromosoma 2002, 110, 550–558. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Verstraete, B.; Groeninckx, I.; Smets, E.; Huysmans, S. Phylogenetic signal of orbicules at family level: Rubiaceae as case study. Taxon 2011, 60, 742–757. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasnyanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003, 10.3.1–10.3.18. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Bohme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Gurusaran, M.; Ravella, D.; Sekar, K. RepEx: Repeat extractor for biological sequences. Genomics 2013, 102, 403–408. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.J.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Swofford, D.L.; Documentation, B. Phylogenetic Analysis Using Parsimony; Illinois Natural History Survey: Champaign, IL, USA, 1989. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1. 4. Molecular Evolution, Phylogenetics and Epidemiology. University of Edinburgh, Institute of Evolutionary Biology: Edinburgh, UK, 2012. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 4 July 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | S. hakusanensis | S. officinalis | S. stipulata | S. tenuifolia |
|---|---|---|---|---|
| Floral characteristics | ||||
| Outer sepal cell surface | Smooth | Smooth | Smooth | Smooth |
| Outer sepal waxes | Platelets | Absent | Absent | Absent |
| Inner sepal cell surface | Striation | Striation | Striation | Striation |
| Filament length | 6–10 mm | 1–3 mm | 5–7 mm | 6–8 mm |
| Filament shape | Compressed-dilated in the upper part | Filiform | Compressed-dilated in the upper part | Compressed-dilated in the upper part |
| Hypanthium shape | Ellipsoid | Ellipsoid | Globose | Globose |
| Disk-like structure * | Moderate | Thickened | Moderate | Reduced |
| Hypanthium cell surface | Smooth | Smooth | Smooth | Smooth |
| Hypanthium trichomes | Basal and ventral suture | Upper part | Absent | Upper part |
| Style length | 2–3 mm | 0.5–1 mm | 1–2 mm | 1–2 mm |
| Style surface | Striation | Striation | Striation | Striation |
| Stigma shape | Fimbriate with blunted apex | Fimbriate with blunted apex | Fimbriate with acute apex | Fimbriate with acute apex |
| Stigma surface | Striation | Striation | Striation | Striation |
| Palynological characteristics | ||||
| Aperture shape | Tricolporate | Hexacolporate | Hexacolporate | Hexacolporate |
| Polar axis (P) | 23.6 ± 1.5 | 16.3 ± 0.8 | 21.6 ± 1.2 | 25.0 ± 1.3 |
| Equatorial diameter (E) | 21.0 ± 1.3 | 15.5 ± 1.1 | 19.6 ± 0.9 | 23.5 ± 1.6 |
| P/E ratio | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 |
| Shape | Prolate-spheroidal | Prolate-spheroidal | Prolate-spheroidal | Prolate-spheroidal |
| Colpus width (C) | 6.7 ± 1.0 | 5.8 ± 0.7 | 7.3 ± 0.9 | 8.7 ± 0.9 |
| Mesocolpus width (M) | 11.3 ± 1.3 | 6.0 ± 0.6 | 7.1 ± 0.9 | 9.4 ± 1.2 |
| C/M ratio | 0.6 ± 0.1 | 1.0 ± 0.2 | 1.0 ± 0.2 | 0.9 ± 0.1 |
| Exine ornamentation | Microechinate | Microechinate | Microechinate | Microechinate |
| Orbicule | Absent | Absent | Absent | Absent |
| Species | S. officinalis | S. stipulata | S. hakusanensis | S. tenuifolia | S. sitchensis | S. filiformis |
|---|---|---|---|---|---|---|
| Accession number | In this study | In this study | In this study | MH513641 | NC_044691 | NC_044693 |
| Total cp genome size (bp) | 155,412 | 155,403 | 155,645 | 155,403 | 155,127 | 154,282 |
| Large single-copy (LSC) region (bp) | 85,478 | 85,444 | 85,697 | 85,525 | 85,347 | 84,405 |
| Inverted repeat (IR) region (bp) | 25,573 | 25,562 | 25,593 | 25,576 | 25,615 | 25,609 |
| Small single-copy (SSC) region (bp) | 18,788 | 18,760 | 18,762 | 18,726 | 18,550 | 18,659 |
| Total number of genes (unique) | 112 | 112 | 112 | 112 | 112 | 112 |
| Protein-coding gene (unique) | 78 | 78 | 78 | 78 | 78 | 78 |
| rRNA (unique) | 4 | 4 | 4 | 4 | 4 | 4 |
| tRNA (unique) | 30 | 30 | 30 | 30 | 30 | 30 |
| GC content (%) | 37.2 | 37.2 | 37.2 | 37.2 | 37.2 | 37.3 |
| LSC (%) | 35.2 | 35.2 | 35.2 | 35.2 | 35.2 | 35.3 |
| IR (%) | 42.7 | 42.7 | 42.7 | 42.7 | 42.7 | 42.8 |
| SSC (%) | 31.2 | 31.2 | 31.3 | 31.3 | 31.4 | 31.4 |
| Species | Region | Marke Name | Aligned Length (bp) | Parsimony Informative Site | Nucleotides Diversity (Pi) | Length of Indel | Number of Haplotype | |
|---|---|---|---|---|---|---|---|---|
| Number | % | |||||||
| Sanguisorba * | matK | S_matK | 689 | 6 | 0.9% | 0.00296 | 0 | 3 |
| rbcL | S_rbcL | 870 | 7 | 0.8% | 0.00200 | 0 | 3 | |
| matK-rps16 | MR16 | 409 | 5 | 1.2% | 0.00466 | 22 | 2 | |
| ycf1 | Y1-1 | 214 | 5 | 2.3% | 0.00699 | 0 | 2 | |
| ycf1 | Y1-2 | 346 | 0 | 0.0% | 0 | 12 | 1 | |
| ycf3 intron | Y3 | 346 | 0 | 0.0% | 0 | 42 | 1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, I.; Song, J.; Yang, S.; Choi, G.; Moon, B. A Comprehensive Study of the Genus Sanguisorba (Rosaceae) Based on the Floral Micromorphology, Palynology, and Plastome Analysis. Genes 2021, 12, 1764. https://doi.org/10.3390/genes12111764
Park I, Song J, Yang S, Choi G, Moon B. A Comprehensive Study of the Genus Sanguisorba (Rosaceae) Based on the Floral Micromorphology, Palynology, and Plastome Analysis. Genes. 2021; 12(11):1764. https://doi.org/10.3390/genes12111764
Chicago/Turabian StylePark, Inkyu, Junho Song, Sungyu Yang, Goya Choi, and Byeongcheol Moon. 2021. "A Comprehensive Study of the Genus Sanguisorba (Rosaceae) Based on the Floral Micromorphology, Palynology, and Plastome Analysis" Genes 12, no. 11: 1764. https://doi.org/10.3390/genes12111764
APA StylePark, I., Song, J., Yang, S., Choi, G., & Moon, B. (2021). A Comprehensive Study of the Genus Sanguisorba (Rosaceae) Based on the Floral Micromorphology, Palynology, and Plastome Analysis. Genes, 12(11), 1764. https://doi.org/10.3390/genes12111764