
A Missense Variant in the Bardet-Biedl Syndrome 2 Gene (BBS2) Leads to a Novel Syndromic Retinal Degeneration in the Shetland Sheepdog

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Whole-Genome Sequencing (WGS)
2.3. Variant Filtering
2.4. Extracting All Variants in BBS Genes
2.5. Variant Validation and Genotyping
3. Results
3.1. Phenotypes
3.2. WGS Variant Filtering
3.3. Variant Mining
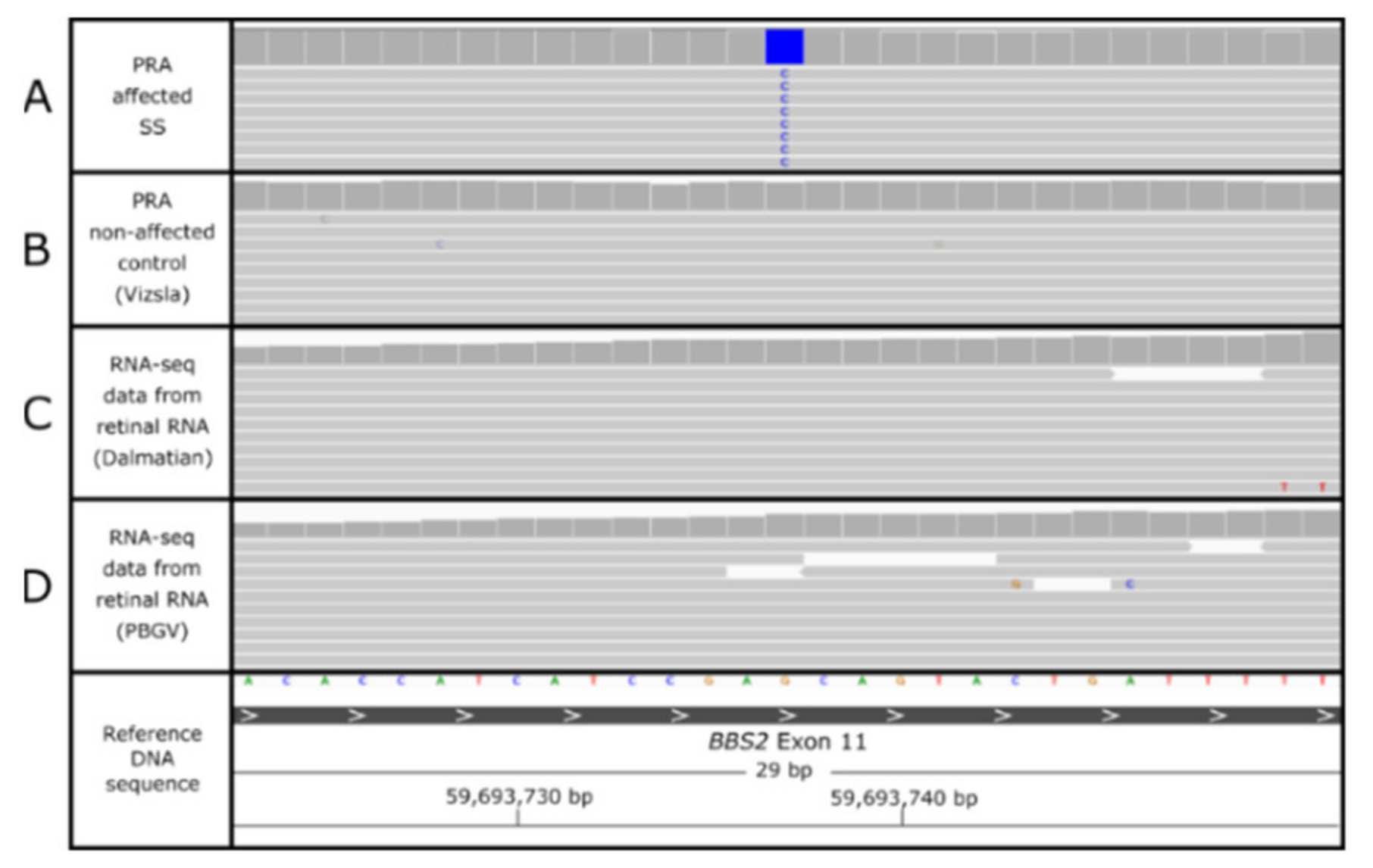
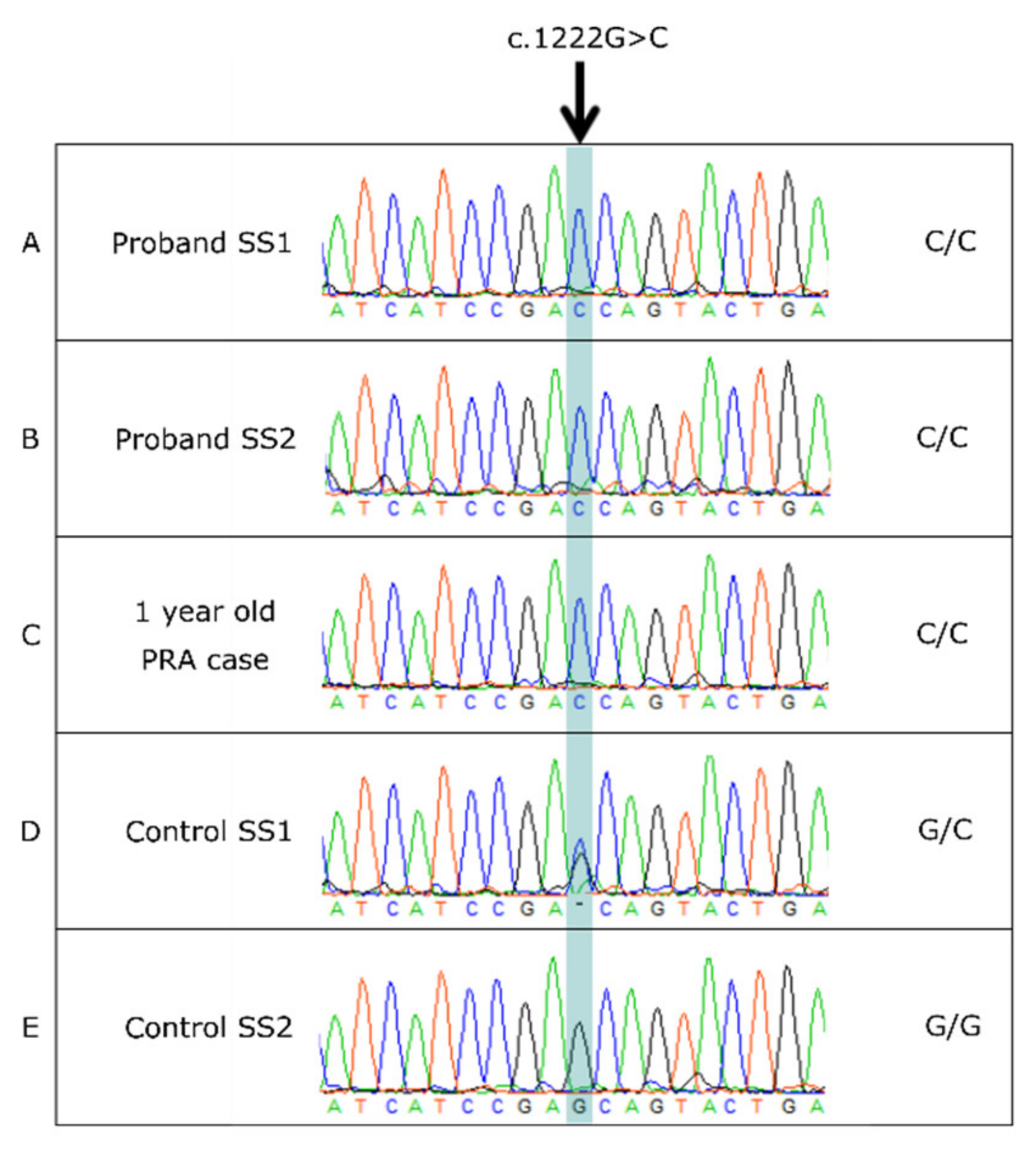
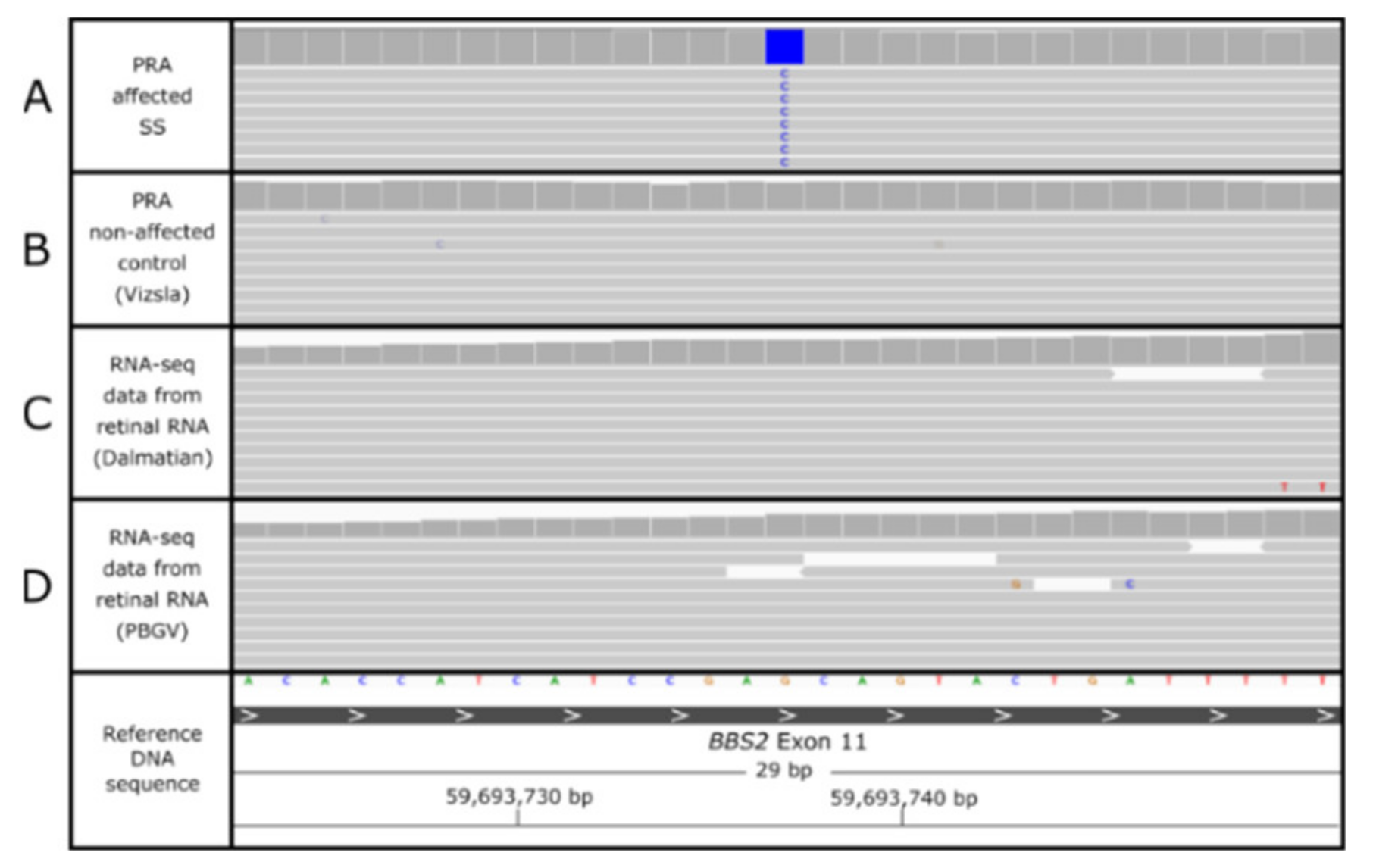
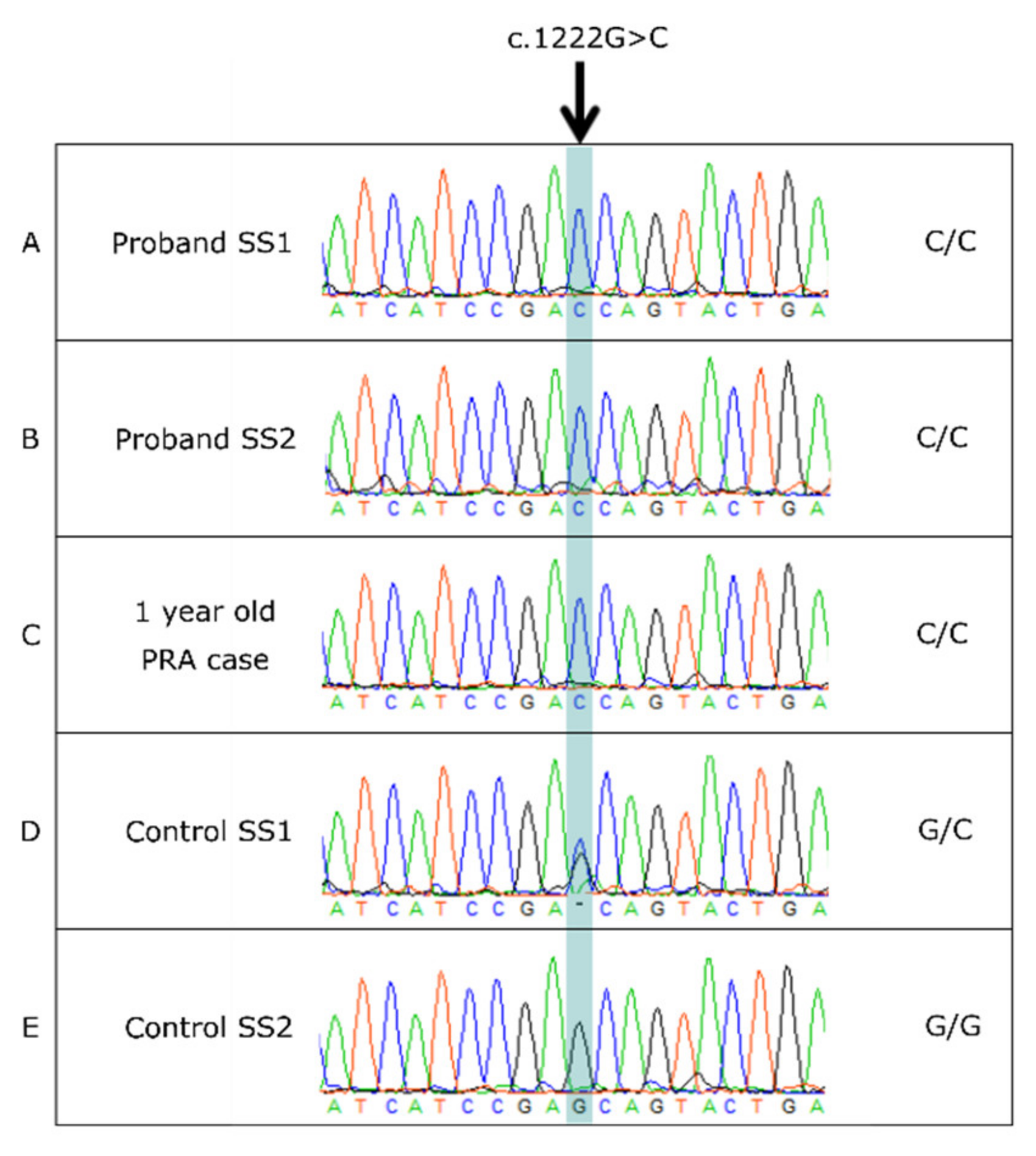
3.4. BBS2 c.1222G>C Missense Variant
3.5. BBS2 c.1222G>C Population Screening
3.6. Characterization of the BBS2-PRA Phenotype
3.7. Additional BBS Gene Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Méndez-Vidal, C.; Bravo-Gil, N.; Pozo, M.G.-D.; Vela-Boza, A.; Dopazo, J.; Borrego, S.; Antiñolo, G. Novel RP1 mutations and a recurrent BBS1variant explain the co-existence of two distinct retinal phenotypes in the same pedigree. BMC Genet. 2014, 15, 9355. [Google Scholar] [CrossRef] [Green Version]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef]
- Mowat, F.M.; Occelli, L.M.; Bartoe, J.T.; Gervais, K.J.; Bruewer, A.R.; Querubin, J.; Dinculescu, A.; Boye, S.L.; Hauswirth, W.; Petersen-Jones, S.M. Gene Therapy in a Large Animal Model of PDE6A-Retinitis Pigmentosa. Front. Neurosci. 2017, 11, 342. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Lheriteau, E.; Weber, M.; Le Meur, G.; Deschamps, J.Y.; Provost, N. Restoration of vision in the pde6beta-deficient dog, a large animal model of rod-cone dystrophy. Mol. Ther. 2012, 20, 2019–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RetNet. RetNet the Retinal Information Network 2021. Available online: http://www.sph.uth.tmc.edu/RetNet/ (accessed on 20 June 2021).
- Downs, L.M.; Wallin-Håkansson, B.; Bergström, T.; Mellersh, C.S. A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever. Canine Genet. Epidemiol. 2014, 1, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, T.; Haase, B.; Bathgate, R.; Willet, C.E.; Kaukonen, M.; Mascord, L.J.; Lohi, H.T.; Wade, C.M. A Coding Variant in the Gene Bardet-Biedl Syndrome 4 (BBS4) Is Associated with a Novel Form of Canine Progressive Retinal Atrophy. G3 Genes Genomes Genet. 2017, 7, 2327–2335. [Google Scholar] [CrossRef]
- Downs, L.M.; Mellersh, C.S. An Intronic SINE Insertion in FAM161A that Causes Exon-Skipping Is Associated with Progressive Retinal Atrophy in Tibetan Spaniels and Tibetan Terriers. PLoS ONE 2014, 9, e93990. [Google Scholar] [CrossRef]
- Kaukonen, M.; Quintero, I.B.; Mukarram, A.K.; Hytönen, M.K.; Holopainen, S.; Wickström, K.; Kyöstilä, K.; Arumilli, M.; Jalomäki, S.; Daub, C.O.; et al. A putative silencer variant in a spontaneous canine model of retinitis pigmentosa. PLoS Genetics 2020, 16, e1008659. [Google Scholar] [CrossRef] [Green Version]
- Hitti-Malin, R.J.; Burmeister, L.M.; Ricketts, S.L.; Lewis, T.W.; Pettitt, L.; Boursnell, M.; Schofield, E.C.; Sargan, D.; Mellersh, C.S. A LINE-1 insertion situated in the promoter of IMPG2 is associated with autosomal recessive progressive retinal atrophy in Lhasa Apso dogs. BMC Genet. 2020, 21, 100. [Google Scholar] [CrossRef]
- Wiik, A.C.; Ropstad, E.O.; Ekesten, B.; Karlstam, L.; Wade, C.M.; Lingaas, F. Progressive retinal atrophy in Shetland sheepdog is associated with a mutation in theCNGA1gene. Anim. Genet. 2015, 46, 515–521. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11. [Google Scholar]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Analyzer NM. 2017. Available online: https://www.ncbi.nlm.nih.gov/Class/Structure/aa/aa_explorer.cgi?mode=translate (accessed on 20 June 2021).
- Adzhubei, I.; Jordan, D.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Staden, R.; Beal, K.F.; Bonfield, J.K. The Staden Package, 1998. Bioinform. Methods Protoc. 2000, 132, 115–130. [Google Scholar] [CrossRef]
- Zhang, Q.; Acland, G.M.; Wu, W.X.; Johnson, J.L.; Pearce-Kelling, S.; Tulloch, B.; Vervoort, R.; Wright, A.F.; Aguirre, G.D. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum. Mol. Genet. 2002, 11, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.S.; Wright, A.F.; Clayton, J.F.; Price, W.H.; Phillips, C.I.; McKeown, C.M.E.; Jay, M.; Bird, A.C.; Pearson, P.L.; Southern, E.M.; et al. Close genetic linkage between X-linked retinitis pigmentosa and a restriction fragment length polymorphism identified by recombinant DNA probe L1.28. Nat. Cell Biol. 1984, 309, 253–255. [Google Scholar] [CrossRef]
- Hardcastle, A.; Thiselton, D.L.; Van Maldergem, L.; Saha, B.K.; Jay, M.; Plant, C.; Taylor, R.; Bird, A.C.; Bhattacharya, S. Mutations in the RP2 Gene Cause Disease in 10% of Families with Familial X-Linked Retinitis Pigmentosa Assessed in This Study. Am. J. Hum. Genet. 1999, 64, 1210–1215. [Google Scholar] [CrossRef] [Green Version]
- Coene, K.; Roepman, R.; Doherty, D.; Afroze, B.; Kroes, H.Y.; Letteboer, S.J.; Ngu, L.H.; Budny, B.; van Wijk, E.; Gorden, N.T.; et al. OFD1 Is Mutated in X-Linked Joubert Syndrome and Interacts with LCA5-Encoded Lebercilin. Am. J. Hum. Genet. 2009, 85, 465–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardcastle, A.; Thiselton, D.L.; Zito, I.; Ebenezer, N.; Mah, T.S.; Gorin, M.B.; Bhattacharya, S.S. Evidence for a new locus for X-linked retinitis pigmentosa (RP23). Investig. Ophthalmol. Vis. Sci. 2000, 41, 2080–2086. [Google Scholar]
- Webb, T.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; May, M.; Tranebjærg, L.; Kendall, E.; Fontán, G.; Jackson, J. A novel X–linked gene, DDP, shows mutations in families with deafness (DFN–1), dystonia, mental deficiency and blindness. Nat. Genet. 1996, 14, 177–180. [Google Scholar] [CrossRef]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Pearce, W.G.; Koop, B.; Fishman, G.A.; Mets, M.; Musarella, M.A.; Boycott, K.M. Loss-of-function mutations in a calcium-channel α1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 264–267. [Google Scholar] [CrossRef]
- Strom, T.M.; Nyakatura, G.; Apfelstedt-Sylla, E.; Hellebrand, H.; Lorenz, B.; Weber, B.H.F. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 260–263. [Google Scholar] [CrossRef]
- Bech-Hansen, N.; Naylor, M.J.; Maybaum, T.A.; Sparkes, R.L.; Koop, B.; Birch, D. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000, 26, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Schinzel, A.; Orth, U.; Fraser, N.A.; Mollica, F.; Craig, I.W.; Kruse, T.; Mächler, M.; Neugebauer, M.; Bleeker-Wagemakers, L.M. Gene of X-chromosomal congenital stationary night blindness is closely linked to DXS7 on Xp. Qual. Life Res. 1989, 81, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, A.; David-Gray, Z.K.; Jay, M.; Bird, A.C.; Bhattacharya, S.S. Localization of CSNBX (CSNB4) between the retinitis pigmentosa loci RP2 and RP3 on proximal Xp. Investig. Ophthalmol. Vis. Sci. 1997, 38, 2750–2755. [Google Scholar]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef] [Green Version]
- Forman, O.P.; Pettitt, L.; Komáromy, A.M.; Bedford, P.; Mellersh, C. A Novel Genome-Wide Association Study Approach Using Genotyping by Exome Sequencing Leads to the Identification of a Primary Open Angle Glaucoma Associated Inversion Disrupting ADAMTS17. PLoS ONE 2015, 10, e0143546. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, E.; Beales, P.L. Bardet–Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl Syndrome-Now and in the Future. Front. Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Niederlova, V.; Modrak, M.; Tsyklauri, O.; Huranova, M.; Stepanek, O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 2019, 40, 2068–2087. [Google Scholar] [CrossRef]
- Loktev, A.V.; Zhang, Q.; Beck, J.S.; Searby, C.C.; Scheetz, T.E.; Bazan, J.F.; Slusarski, D.C.; Sheffield, V.C.; Jackson, P.K.; Nachury, M.V. A BBSome Subunit Links Ciliogenesis, Microtubule Stability, and Acetylation. Dev. Cell 2008, 15, 854–865. [Google Scholar] [CrossRef] [Green Version]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peränen, J.; Merdes, A. A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yu, D.; Seo, S.; Stone, E.M.; Sheffield, V.C. Intrinsic Protein-Protein Interaction-mediated and Chaperonin-assisted Sequential Assembly of Stable Bardet-Biedl Syndrome Protein Complex, the BBSome. J. Biol. Chem. 2012, 287, 20625–20635. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The Conserved Bardet-Biedl Syndrome Proteins Assemble a Coat that Traffics Membrane Proteins to Cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef] [Green Version]
- Ludlam, W.G.; Aoba, T.; Cuéllar, J.; Bueno-Carrasco, M.T.; Makaju, A.; Moody, J.D.; Franklin, S.; Valpuesta, J.M.; Willardson, B.M. Molecular architecture of the Bardet–Biedl syndrome protein 2-7-9 subcomplex. J. Biol. Chem. 2019, 294, 16385–16399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Bee, Y.M.; Chawla, M.; Zhao, Y. Whole Exome Sequencing Identifies a Novel and a Recurrent Mutation inBBS2Gene in a Family with Bardet-Biedl Syndrome. BioMed Res. Int. 2015, 2015, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ece Solmaz, A.; Onay, H.; Atik, T.; Aykut, A.; Cerrah Gunes, M.; Ozalp Yuregir, O.; Bas, V.N.; Hazan, F.; Kirbiyik, O.; Ozkinay, F. Targeted multi-gene panel testing for the diagnosis of Bardet Biedl syndrome: Identification of nine novel mutations across BBS1, BBS2, BBS4, BBS7, BBS9, BBS10 genes. Eur. J. Med Genet. 2015, 58, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D.Y.; Searby, C.C.; Carmi, R.; Elbedour, K.; Van Maldergem, L.; Fulton, A.B.; Lam, B.L.; Powell, B.R.; Swiderski, R.E.; Bugge, K.E.; et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2). Hum. Mol. Genet. 2001, 10, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Shevach, E.; Ali, M.; Mizrahi-Meissonnier, L.; McKibbin, M.; El-Asrag, M.; Watson, C.M.; Inglehearn, C.F.; Ben-Yosef, T.; Blumenfeld, A.; Jalas, C.; et al. Association Between Missense Mutations in theBBS2Gene and Nonsyndromic Retinitis Pigmentosa. JAMA Ophthalmol. 2015, 133, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, D.Y.; Fath, M.; Mullins, R.F.; Searby, C.; Andrews, M.; Davis, R.; Andorf, J.L.; Mykytyn, K.; Swiderski, R.E.; Yang, B.; et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 16588–16593. [Google Scholar] [CrossRef] [Green Version]
- Yen, H.J. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer’s vesicle cilia function. Hum. Mol. Genet. 2006, 15, 667–677. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Smaoui, N.; Hammer, M.B.H.; Jiao, X.; Riazuddin, S.A.; Harper, S.; Katsanis, N.; Riazuddin, S.; Chaabouni, H.; Berson, E.L.; et al. Molecular Analysis of Bardet-Biedl Syndrome Families: Report of 21 Novel Mutations in 10 Genes. Investig. Opthalmol. Vis. Sci. 2011, 52, 5317–5324. [Google Scholar] [CrossRef] [PubMed]
- Bin, J.; Madhavan, J.; Ferrini, W.; Mok, C.A.; Billingsley, G.; Héon, E. BBS7andTTC8(BBS8) mutations play a minor role in the mutational load of Bardet-Biedl syndrome in a multiethnic population. Hum. Mutat. 2009, 30, E737–E746. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic Inheritance in Bardet-Biedl Syndrome, a Mendelian Recessive Disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorda-Sanchez, I.; Ayuso, C.; Sanz, R.; Ibañez, A. Does Bardet-Biedl syndrome have a characteristic face? J. Med. Genet. 2001, 38, 14. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.J.; Green, J.S.; Fan, Y.; Bhogal, A.K.; Dicks, E.; Fernandez, B.A.; Stefanelli, M.; Murphy, C.; Cramer, B.C.; Dean, J.C.; et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: A 22-year prospective, population-based, cohort study. Am. J. Med Genet. Part A 2005, 132A, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.E.; Swiderski, R.E.; Rahmouni, K.; Nishimura, D.Y.; Mullins, R.; Agassandian, K.; Philp, A.R.; Searby, C.C.; Andrews, M.P.; Thompson, S.; et al. A knockin mouse model of the Bardet Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 19422–19427. [Google Scholar] [CrossRef] [Green Version]
- Rahmouni, K.; Fath, M.; Seo, S.; Thedens, D.R.; Berry, C.J.; Weiss, R.; Nishimura, D.Y.; Sheffield, V. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome. J. Clin. Investig. 2008, 118, 1458–1467. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.; Parker, D.; Flinter, F. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med Genet. 1999, 36, 437–446. [Google Scholar]
- Feuillan, P.P.; Ng, D.; Han, J.C.; Sapp, J.; Wetsch, K.; Spaulding, E.; Zheng, Y.C.; Caruso, R.C.; Brooks, B.P.; Johnston, J.J.; et al. Patients with Bardet-Biedl Syndrome Have Hyperleptinemia Suggestive of Leptin Resistance. J. Clin. Endocrinol. Metab. 2011, 96, E528–E535. [Google Scholar] [CrossRef]
- Imhoff, O.; Marion, V.; Stoetzel, C.; Durand, M.; Holder, M.; Sigaudy, S.; Sarda, P.; Hamel, C.P.; Brandt, C.; Dollfus, H.; et al. Bardet-Biedl Syndrome: A Study of the Renal and Cardiovascular Phenotypes in a French Cohort. Clin. J. Am. Soc. Nephrol. 2010, 6, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, E.; Sparks, K.; Best, S.; Borrows, S.; Hoskins, B.; Sabir, A.; Barrett, T.; Williams, D.; Mohammed, S.; Goldsmith, D.; et al. Risk Factors for Severe Renal Disease in Bardet–Biedl Syndrome. J. Am. Soc. Nephrol. 2016, 28, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.M.; Michaud, E.J.; Schoeb, T.R.; Aydin-Son, Y.; Miller, M.; Yoder, B.K. The Oak Ridge Polycystic Kidney mouse: Modeling ciliopathies of mice and men. Dev. Dyn. 2008, 237, 1960–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkeläinen, S.; Hellsand, M.; van der Heiden, A.D.; Andersson, E.; Thorsson, E.; Ström-Holst, B.; Häggström, J.; Ljungvall, I.; Mellersh, C.; Hallböök, F.; et al. Deletion in the Bardet-Biedl syndrome gene TTC8 results in a syndromic retinal degeneration in dogs. Genes 2020, 11, 1090. [Google Scholar] [CrossRef]
- Courcier, E.A.; Thomson, R.M.; Mellor, D.J.; Yam, P.S. An epidemiological study of environmental factors associated with canine obesity. J. Small Anim. Pract. 2010, 51, 362–367. [Google Scholar] [CrossRef]
- German, A.J.; Woods, G.R.T.; Holden, S.L.; Brennan, L.; Burke, C. Dangerous trends in pet obesity. Veter. Rec. 2018, 182, 25. [Google Scholar] [CrossRef] [Green Version]
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Alfadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Cohort | c.1222G>C C/C | c.1222G>C G/C | c.1222G>C G/G | Total SS Genotyped |
|---|---|---|---|---|
| AHT | 5 | 11 | 75 | 91 |
| The University of Helsinki | 0 | 6 | 388 | 394 |
| The Norwegian University of Life Sciences | 2 | 0 | 6 | 8 |
| The University of Pennsylvania | 0 | 0 | 9 | 9 |
| DBVDC | 0 | 0 | 3 | 3 |
| Total SS genotyped | 505 |
| Confirmed PRA Cases | PRA Non-Affected | Unknown PRA Diagnosis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| c.1222G>C genotype | C/C | G/C | G/G | C/C | G/C | G/G | C/C | G/C | G/G |
| Number of individuals | 4 | 0 | 10 | 2 † | 11 | 29 | 1 | 6 | 442 |
| Breed | c.1222G>C Genotype | Allele Frequency of Causal Allele | ||
|---|---|---|---|---|
| C/C | G/C | G/G | ||
| SS | 7 | 17 | 481 | 0.031 |
| SS * | 4 | 13 | 481 | 0.021 |
| Crossbreeds | 0 | 0 | 15 | 0 |
| Wolves | 0 | 0 | 8 | 0 |
| 154 other breeds | 0 | 0 | 858 | 0 |
| Proband | Ocular Phenotype | Age of Diagnosis | Age at Submission | Additional Phenotypic Features |
|---|---|---|---|---|
| SS1 | PRA confirmed | 8.7 years | 9 years |
|
| SS2 | PRA confirmed | 6 years | 9 years |
|
| SS3 | PRA unconfirmed but blind | 9–10 years | 9 years |
|
| SS4 | No ophthalmoscopic examination | N/A | 1 year |
|
| SS5 | PRA unaffected | 7.6 years | 7.6 years |
|
| SS6 | PRA confirmed | Unknown | Unknown |
|
| SS7 | PRA confirmed | Unknown | Unknown |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hitti-Malin, R.J.; Burmeister, L.M.; Lingaas, F.; Kaukonen, M.; Pettinen, I.; Lohi, H.; Sargan, D.; Mellersh, C.S. A Missense Variant in the Bardet-Biedl Syndrome 2 Gene (BBS2) Leads to a Novel Syndromic Retinal Degeneration in the Shetland Sheepdog. Genes 2021, 12, 1771. https://doi.org/10.3390/genes12111771
Hitti-Malin RJ, Burmeister LM, Lingaas F, Kaukonen M, Pettinen I, Lohi H, Sargan D, Mellersh CS. A Missense Variant in the Bardet-Biedl Syndrome 2 Gene (BBS2) Leads to a Novel Syndromic Retinal Degeneration in the Shetland Sheepdog. Genes. 2021; 12(11):1771. https://doi.org/10.3390/genes12111771
Chicago/Turabian StyleHitti-Malin, Rebekkah J., Louise M. Burmeister, Frode Lingaas, Maria Kaukonen, Inka Pettinen, Hannes Lohi, David Sargan, and Cathryn S. Mellersh. 2021. "A Missense Variant in the Bardet-Biedl Syndrome 2 Gene (BBS2) Leads to a Novel Syndromic Retinal Degeneration in the Shetland Sheepdog" Genes 12, no. 11: 1771. https://doi.org/10.3390/genes12111771
APA StyleHitti-Malin, R. J., Burmeister, L. M., Lingaas, F., Kaukonen, M., Pettinen, I., Lohi, H., Sargan, D., & Mellersh, C. S. (2021). A Missense Variant in the Bardet-Biedl Syndrome 2 Gene (BBS2) Leads to a Novel Syndromic Retinal Degeneration in the Shetland Sheepdog. Genes, 12(11), 1771. https://doi.org/10.3390/genes12111771